Good Manufacturing Practice, or GMP, is the universal standard for quality production. It is a set of rules to ensure that products like medicines, food, and medical devices are made consistently and safely, batch after batch. The central idea is simple: quality cannot be inspected into a product at the end of the line. Instead, it must be built into every step of the manufacturing process, from the raw materials that arrive at the loading dock to the final package that leaves it.
The “c” in cGMP stands for Current. This single letter introduces a critical, dynamic requirement. While GMP provides the foundational rulebook, cGMP legally obligates manufacturers to use the most up-to-date technologies, systems, and scientific understanding available today. A process that was perfectly acceptable under GMP standards a decade ago might fail a cGMP inspection now if better, more reliable methods have since emerged. It forces companies to continuously improve.
Key Takeaways

- GMP & cGMP distinction is now academic; the expectation is universal.
- Quality Risk Management (QRM) is the engine, not the paperwork nor the PLM.
- Data integrity is a primary audit focus.
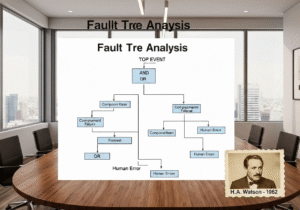
- “Human Error” is a symptom, not a root cause. Attributing a deviation to “human error” is a red flag for a weak quality system.
- Process Analytical Technology (PAT) embodies the shift from testing to real-time assurance. The “c” in cGMP is exemplified by PAT.
- Supplier oversight is data-driven, not just audit-driven.
- The Qualified Person (QP) represents a critical EU-specific responsibility.
- The Contamination Control Strategy (CCS) is the new cornerstone of sterile manufacturing.
The 10 Core Principles of Good Manufacturing Practice (GMP)
GMP is not just a set of rules but a quality mindset built upon ten fundamental principles. These principles work together to create a robust system that ensures quality is built into a product at every stage, rather than merely being tested for at the end.

1. Write Step-by-Step Procedures and Work Instructions
The foundation of GMP is ensuring that all processes are clearly defined and documented. This principle requires creating detailed, unambiguous Standard Operating Procedures (SOPs) for every critical task. The goal is to ensure that operations are performed consistently and correctly every time, regardless of who is performing the task. This eliminates ambiguity and provides a clear reference for training and execution.
Example of application: a company, “PharmaBlend Inc.,” manufactures a temperature-sensitive liquid drug. Their SOP for “Compounding Tank Temperature Control” (SOP-MFG-101) specifies not just the target temperature (40°C ± 2°C), but also the exact sequence for starting the heating jacket, the rate of temperature increase (not to exceed 5°C per minute), the specific calibrated probe to use for monitoring, and the actions to take if the temperature overshoots.
Tip: instead of writing monolithic SOPs, use a modular approach. Create “master” SOPs for complex processes that reference smaller, task-specific “work instruction” documents for individual steps (e.g., calibrating a specific sensor, operating a single valve). This allows for easier updates—if a single piece of equipment is replaced, you only need to revise one small work instruction instead of the entire process SOP, significantly reducing review and approval time and minimizing the risk of introducing errors into unrelated sections.
2. Follow Procedures and Instructions Meticulously

Having documented procedures is meaningless if they are not followed. This principle demands strict adherence to the written SOPs without deviation. If a deviation is necessary, it must be formally documented, justified, and approved through a defined change control process. This ensures that any departure from the standard is controlled, assessed for risk, and recorded for traceability.
Tip: implement a “Right-First-Time” (RFT) metric for procedure execution, tracked during batch record review. When deviations occur due to non-adherence, don’t just retrain the operator. Perform a root cause analysis focused on the procedure’s usability (a Human Factors approach). Was the instruction ambiguous? Was the sequence illogical? Is the required tool hard to access? Improving the procedure itself is a more effective long-term Corrective and Preventive Action (CAPA) than simply blaming human error.
3. Promptly and Accurately Document Work

This is the principle of “if it wasn’t written down, it didn’t happen.” All activities, from receiving raw materials to shipping the final product, must be documented in real-time. This includes recording data, signatures, dates, and any observations. Accurate, contemporaneous documentation provides a complete and traceable history of a batch (known as a Batch Record or Device History Record), which is essential for investigating deviations, troubleshooting problems, and proving compliance during an audit.
Tip: when designing batch records (paper or electronic), incorporate “data integrity checks” directly into the fields. For example, instead of just a blank space for “End Time,” structure it to require a start time and an end time, with an automated or manual check to ensure the duration is logical for the process step. For critical entries, use “verify-by-second-person” sign-offs, but ensure the verifier is trained to re-perform the critical calculation or check the setting, not just “check the box.”
Difference between DMR and DHR:
- The Device Master Record, or DMR, is the master recipe for a medical device. It is a formal, controlled compilation of all the instructions, specifications, and procedures required to produce a consistent product. The DMR contains everything from the design drawings and material specifications to the detailed manufacturing instructions, quality control test methods, labeling, and packaging requirements. Think of it as the complete blueprint; it defines exactly how the device is supposed to be made, from start to finish.
- The Device History Record, or DHR, is the proof that a specific batch, lot, or individual unit was actually built according to that recipe. It is the completed production record. The DHR contains the specific dates of manufacture, quantities produced, test results for that batch, and traceability information like serial or lot numbers. While the DMR is the instruction manual that applies to all units, the DHR is the historical evidence that demonstrates one specific production run followed those instructions and met all acceptance criteria.
4. Validate Your Work & Process

Validation is the documented proof that a process, system, or piece of equipment consistently produces the expected result. This principle requires manufacturers to prove that their processes are reliable...
You have read 14% of the article. The rest is for our community. Already a member? Log in
(and also to protect our original content from scraping bots)
Innovation.world community
Login or Register (100% free)
View the rest of this article and all members-only content and tools.
Only real engineers, manufacturers, designers, marketers professionals.
No bot, no hater, no spammer.
FAQ
In a practical audit, how does an inspector’s expectation for ‘cGMP’ differ from the written ‘GMP’ regulations?
An inspector expects to see not just that you follow your written procedures (GMP), but that your procedures themselves reflect current industry best practices and technology (cGMP). They will question why you are using a 20-year-old analytical method when a more accurate and reliable one is now standard, or why you rely on manual checks where automated in-line verification is now common. They are auditing your awareness and proactive adoption of modern quality standards.
Does cGMP mean we must constantly invest in the newest technology, or can we justify using older, validated equipment?
You can absolutely justify using older equipment, but the burden of proof is on you. Your justification must be documented and risk-based. You need to demonstrate through robust validation, rigorous maintenance, intensive monitoring, and trend data that your older system provides an equivalent or superior level of quality assurance and process control compared to modern alternatives. If your process using old equipment has a higher deviation rate, you will not be able to defend it.
Beyond audit trails, what are the most common ‘unseen’ data integrity gaps regulators are focusing on?
Regulators are heavily scrutinizing uncontrolled spreadsheets used for GMP calculations, the use of shared login credentials on standalone equipment (like balances or pH meters), and the ability to perform “test runs” on analytical equipment that can be deleted without a trace. Another major focus is the integrity of metadata—the data about the data, such as timestamps and user IDs, which must be securely linked to the original record.
How is a Pharmaceutical Quality System (PQS) under ICH Q10 different from just having a strong QA department?
A strong QA department enforces quality; a PQS manages it as a business-wide objective. The key difference is the formal integration of senior management and a focus on process performance and continuous improvement. A PQS ensures that quality metrics directly influence business decisions (like resource allocation and strategic planning) and that management is actively reviewing and driving the system’s effectiveness, as opposed to delegating all quality matters to QA.
What does a “lifecycle approach” to process validation (per ASTM E2500) actually mean for an engineer?
It means validation is no longer a “three-and-done” batch exercise. It’s a continuous process. For an engineer, this means: Stage 1 (Process Design): using Quality by Design (QbD) to define a robust process and its control space. Stage 2 (Process Qualification): verifying the facility and equipment are fit for purpose and that the process consistently works within its defined space (PPQ). Stage 3 (Continued Process Verification): actively monitoring the process during routine production using statistical process control (SPC) to ensure it remains in a state of control for its entire commercial life.
What is the most significant practical difference between EU GMP (EudraLex) and US cGMP?
The most significant difference is the role of the Qualified Person (QP) in the EU. In the US, the Quality Unit has the authority to release a batch. In the EU, a specifically named QP must personally certify that each batch has been manufactured and tested in accordance with all regulations and the marketing authorization before it can be released. This places an immense personal and legal responsibility on one individual.
Our CAPA system is compliant, but problems often recur. What is the cGMP expectation for “CAPA effectiveness”?
The cGMP expectation is that you formally verify your CAPAs worked. This requires building an “effectiveness check” step into your CAPA procedure. This check, performed weeks or months after the CAPA is implemented, must provide objective data (e.g., trend analysis of deviation rates, new audit findings) to prove that the root cause was eliminated and the problem has not recurred. A CAPA closed without this verification is a major red flag for auditors.
How has the cGMP expectation for supplier qualification evolved beyond just auditing the supplier?
Audits are still necessary, but cGMP now expects a more data-driven, risk-based approach. This includes establishing formal Quality Agreements that define responsibilities, monitoring the supplier’s performance through metrics (e.g., on-time delivery, deviation rates, quality of incoming material), and performing periodic raw material testing to verify the supplier’s Certificate of Analysis (CoA). You must demonstrate ongoing oversight, not just a one-time qualification.
With the revision of Annex 1, what is the single biggest cGMP shift in sterile manufacturing?
The biggest shift is the mandate for a formal, holistic Contamination Control Strategy (CCS). This is not just a collection of SOPs but a single, comprehensive document that justifies your facility design, processes, and monitoring programs based on risk management. It forces you to demonstrate how all your individual control measures (from gowning to HVAC to process design) work together to prevent contamination.
Why is Process Analytical Technology (PAT) considered a pillar of modern cGMP?
Because PAT embodies the core cGMP principle of building quality in, rather than testing it in. By providing real-time, in-process data, PAT allows for the active control of Critical Process Parameters (CPPs) to ensure Critical Quality Attributes (CQAs) are met. This shifts manufacturing from a rigid, recipe-based approach to a flexible, science-based model that can adjust to minor variabilities and guarantee a consistent outcome.
How should “human error” be treated as a root cause in a cGMP environment?
In a mature cGMP system, “human error” is rarely an acceptable root cause. It is usually a symptom of a flawed process or system. When an error occurs, the investigation must dig deeper: Was the procedure confusing? Was the training inadequate? Was the workspace poorly designed (human factors engineering)? Was the operator fatigued due to excessive overtime? A robust CAPA will address the underlying system failure, not just retrain the individual.
The Annual Product Review (PQR) is often seen as a chore. What is its intended cGMP purpose?
Its intended purpose is to be a proactive tool for continuous improvement. The PQR should not just be a retrospective data dump. It is a formal opportunity to analyze a year’s worth of data (trends, deviations, changes, stability results) to assess the health and consistency of a process. Its most important output should be a list of recommended CAPAs and process improvements for the upcoming year.
Related Readings
External Links on
International Standards
- ISO 22716:2007 Good Manufacturing Practices (GMP) - Guidelines on Good Manufacturing Practices
- FDA 21 CFR Part 210 and 211: Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs
- ISO 13485:2016 Medical devices - Quality management systems - Requirements for regulatory purposes
(hover the link to see our description of the content)
Glossary of Terms Used
American Society for Testing and Materials (ASTM): an international standards organization that develops and publishes voluntary consensus technical standards for materials, products, systems, and services, aimed at improving quality and safety across various industries.
Application Programming Interface (API): a set of rules and protocols that allows different software applications to communicate and interact with each other, enabling the integration of functionalities and data exchange between systems.
Certificate of Analysis (CoA): a document issued by a manufacturer or testing laboratory that confirms a product's specifications, quality, and compliance with regulatory standards, detailing test results and methods used for analysis.
Contamination Control Strategy (CCS): a systematic approach to prevent, detect, and mitigate contamination in controlled environments, ensuring product quality and safety through defined procedures, monitoring, and risk management practices.
Corrective Action and Preventative Action (CAPA): a systematic approach to identifying, investigating, and addressing nonconformities and potential issues to prevent recurrence and ensure compliance with regulatory standards in quality management systems.
Critical Control Points (CCP): specific stages in a process where control can be applied to prevent, eliminate, or reduce food safety hazards to acceptable levels. Identifying these points is essential for effective hazard analysis and critical control management in food production systems.
current Good Manufacturing Practice (cGMP): a system ensuring that products are consistently produced and controlled according to quality standards, encompassing regulations and guidelines for manufacturing processes, facilities, equipment, and personnel to ensure safety, quality, and efficacy in pharmaceuticals, food, and other regulated industries.
Device History Record (DHR): a compilation of records that documents the production history of a medical device, including manufacturing, quality control, and testing data, ensuring compliance with regulatory requirements and facilitating traceability throughout the device's lifecycle.
Device Master Record (DMR): a compilation of documents and specifications that provide the necessary information to produce a medical device, including design specifications, production processes, quality assurance measures, and labeling requirements, ensuring compliance with regulatory standards.
Failure Mode and Effects Analysis (FMEA): a systematic method for evaluating potential failure modes within a system, process, or product, assessing their effects on performance, and prioritizing risks to improve reliability and safety through corrective actions.
Food and Drug Administration (FDA): a federal agency of the United States Department of Health and Human Services responsible for regulating food safety, pharmaceuticals, medical devices, cosmetics, and tobacco products to ensure public health and safety through scientific evaluation and enforcement of compliance standards.
Good Manufacturing Practice (GMP): a system ensuring products are consistently produced and controlled according to quality standards, minimizing risks involved in pharmaceutical production and related industries. It encompasses guidelines for manufacturing processes, facility conditions, personnel qualifications, and documentation practices to ensure product safety and efficacy.
Hazard Analysis and Critical Control Points (HACCP): a systematic approach to food safety that identifies, evaluates, and controls hazards at critical points in the production process to prevent foodborne illnesses and ensure product safety.
Heating Ventilation and Air Conditioning (HVAC): a system designed to regulate indoor climate by controlling temperature, humidity, and air quality through heating, cooling, and ventilation processes. It includes components such as furnaces, air conditioners, ductwork, and thermostats for efficient environmental management.
Installation Qualification (IQ): a documented process to verify that equipment or systems are installed according to specifications, including assessment of utilities, environmental conditions, and compliance with design requirements, ensuring readiness for operational qualification.
International Organization for Standardization (ISO): a non-governmental international body that develops and publishes standards to ensure quality, safety, efficiency, and interoperability across various industries and sectors, facilitating global trade and cooperation. Established in 1947, it comprises national standardization organizations from member countries.
Key Performance Indicator (KPI): a measurable value that demonstrates how effectively an organization is achieving key business objectives, often used to evaluate success at reaching targets.
Magnetic Resonance Imaging (MRI): a medical imaging technique that uses strong magnetic fields and radio waves to generate detailed images of internal body structures, particularly soft tissues, by detecting the signals emitted from hydrogen nuclei in the presence of a magnetic field.
Operational Qualification (OQ): a validation process that ensures equipment or systems operate according to specified requirements within defined limits, confirming that they perform as intended in their operational environment.
parts per million (ppm): a unit of measurement representing the concentration of one substance in one million parts of another, often used to quantify pollutants or contaminants in air, water, or soil. It is equivalent to milligrams of substance per liter of solution or per kilogram of material.
Performance Qualification (PQ): a process that verifies a system or equipment operates according to specified requirements under real-world conditions, ensuring it consistently performs its intended function within predetermined limits.
Product Lifecycle Management (PLM): a systematic approach to managing a product's lifecycle from inception, through engineering design and manufacturing, to service and disposal, integrating people, processes, data, and technology to improve product quality, reduce time to market, and enhance collaboration across stakeholders.
Qualified Person (QP): an individual with the necessary education, experience, and authority to oversee and ensure compliance with regulatory requirements in the preparation and submission of technical documents, particularly in the mining and resource sectors, as defined by relevant industry standards.
Standard Operating Procedure (SOP): a set of step-by-step instructions created to help workers carry out routine operations consistently and efficiently, ensuring compliance with regulations and quality standards.
Statistical Process Control (SPC): a method of quality control that employs statistical techniques to monitor and control a process, ensuring it operates at its full potential by identifying variations and maintaining consistent output within specified limits.