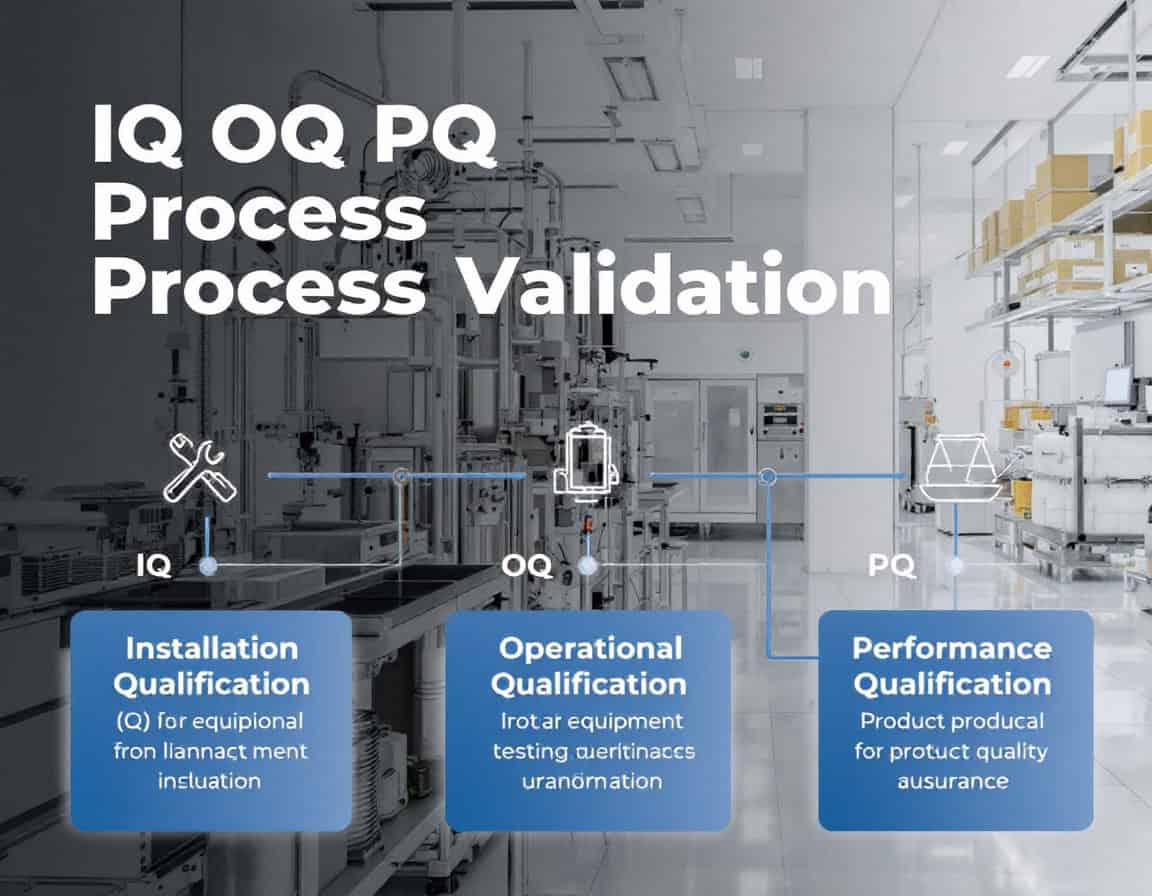
Die IQ OQ PQ-Prozessvalidierungsmethode, die für Installation Qualification (IQ), Operational Qualification (OQ) und Performance Qualification (PQ) steht, stellt die etablierte Rahmen Zur Prozessvalidierung. Sie dient der systematischen Erstellung dokumentierter Nachweise dafür, dass ein Herstellungsprozess unter Kontrolle ist. Diese Methodik folgt einem bewussten und logischen Ablauf. Sie beginnt mit der statischen, dokumentierten Überprüfung der korrekten Installation der Ausrüstung (IQ) und geht über zur dynamischen Bestätigung, dass die Ausrüstung in ihren spezifizierten Bereichen zuverlässig arbeitet (OQ). Abschließend wird nachgewiesen, dass der integrierte Prozess durchgängig Produkte produziert, die alle Qualitätsmerkmale erfüllen (PQ).
Der gesamte Ablauf ist darauf ausgelegt, vor der kommerziellen Nutzung einen objektiven, nachvollziehbaren Nachweis dafür zu erbringen, dass der Prozess robust, reproduzierbar und für den vorgesehenen Zweck geeignet ist.
Diese Methodik ist eine nicht verhandelbare regulatorische Voraussetzung in Branchen, die cGMP unterliegen, wie z. B. Pharmazeutika, Biologika und Medizinprodukt Herstellung, bei der die Prozesskonsistenz direkt mit der Patientensicherheit verknüpft ist. Die Anwendung dient nicht der anfänglichen Forschung, sondern ist die kritische Validierungsphase, die nach Abschluss der Prozessentwicklung und vor der Zulassung des Prozesses für die routinemäßige kommerzielle Produktion durchgeführt wird.
Die wichtigsten Erkenntnisse

- Das Ergebnis der Validierung ist ein Beweis, kein Produkt.
- Beim „Worst-Case“-OQ-Test handelt es sich um kombinierte Stressoren.
- PQ validiert den integrierten Prozess, nicht nur die Ausrüstung.
- Die „Drei-Chargen“-Regel demonstriert die Reproduzierbarkeit.
- Der Erfolg von PQ wird durch Konsistenz definiert, nicht nur durch das Erfüllen von Spezifikationen.
- Das Protokoll ist ein Vertrag; Abweichungen müssen begründet und gut dokumentiert werden.
- Nutzen Sie die Lieferantendokumentation (FAT/SAT), ersetzen Sie jedoch nicht Ihre eigene OQ
- Die Validierung definiert den Beginn des Lebenszyklus, nicht das Ende eines Projekts.
- Jede „gleichwertige“ Änderung bedarf einer Begründung.
- Personalschulung ist Voraussetzung für OQ und PQ
Installationsqualifizierung (IQ)
Die Installationsqualifizierung (IQ) ist die erste und grundlegende Phase der IQ OQ PQ-Prozessvalidierungsmethodik. Es handelt sich um einen formalen, dokumentierten Prozess, der überprüft und bestätigt, dass Geräte, Systeme und Zusatzkomponenten in vollständiger Übereinstimmung mit den Empfehlungen des Herstellers und den Designspezifikationen des Benutzers geliefert, installiert und konfiguriert wurden.
Das Kernprinzip von IQ besteht darin, dokumentierte Nachweise dafür zu erbringen, dass die Installation korrekt ist und eine geeignete und sichere Umgebung für die nachfolgenden Qualifizierungsphasen schafft.
Zweck und Bedeutung des IQ: Das Hauptziel der IQ besteht darin, eine Basis für die Gerätevalidierung zu schaffen. Bevor mit den Betriebstests begonnen werden kann, muss sichergestellt werden, dass die Geräte physisch vorhanden, korrekt montiert und in einer geeigneten Umgebung aufgestellt sind. Diese Phase minimiert die Risiken einer unsachgemäßen Installation, die andernfalls zu Gerätestörungen, Produktqualitätsproblemen und Sicherheitsrisiken führen kann.
Wichtige Phasen und Aktivitäten der Installationsqualifizierung
Der IQ-Prozess ist systematisch und umfasst mehrere detaillierte Phasen, die normalerweise über ein vorab genehmigtes Protokoll und eine Checkliste verwaltet werden.
1. Vorinstallation und Planung

Bevor die Ausrüstung eintrifft oder bestellt wird, beginnt der IQ-Prozess mit einer sorgfältigen Planung. Dazu gehören:
- Überprüfung der Site-Bereitschaft: Stellen Sie sicher, dass der vorgesehene Standort für die neue Ausrüstung vorbereitet ist. Dazu gehört die Überprüfung auf ausreichend Platz, strukturelle Unterstützung und die Verfügbarkeit der erforderlichen Versorgungseinrichtungen an den richtigen Anschlusspunkten.
- Umweltbedingungen: Überprüfen Sie, ob die Installationsumgebung den Herstellerspezifikationen hinsichtlich Temperatur, Luftfeuchtigkeit und Sauberkeit entspricht.
- Dokumentation sammeln: Sammeln Sie alle wichtigen Dokumente, wie z. B. die Bestellung, Herstellerhandbücher, Konstruktionsspezifikationen, technische Zeichnungen und Kalibrierungszertifikate für alle integrierten Messinstrumente.
Tipp: Gehen Sie den Weg ab und modellieren Sie das Volumen. Betrachten Sie nicht nur einen 2D-Grundriss. Hier passieren die häufigsten und kostspieligsten IQ-Fehler. Gehen Sie die geplante Route von der Laderampe bis zum endgültigen Installationsort mit Ihren wichtigsten Beteiligten (Einrichtungen, Technik und dem Projektmanager des Lieferanten) ab. Verwenden Sie einen einfachen Holz- oder PVC-Rahmen, der auf die maximale Höhe, Breite und Länge der Maschine zugeschnitten ist. Tragen Sie diesen „Geisterrahmen“ physisch den gesamten Weg entlang. Dadurch werden Probleme mit der Durchfahrtshöhe bei Türen, tief hängenden Rohren, engen Ecken und Aufzugskapazitäten sichtbar, die in Zeichnungen oft übersehen werden. Überprüfen Sie außerdem die Bodenbelastbarkeit nicht nur am Zielort, sondern entlang des gesamten Transportwegs.

2. Geräteempfang und -überprüfung
Nach der Lieferung der Ausrüstung wird eine gründliche Inspektion durchgeführt:
- Komponentenüberprüfung: Die gelieferten Artikel werden sorgfältig anhand der Packliste und der Bestellung überprüft, um sicherzustellen, dass alle Komponenten, einschließlich der Software, eingegangen sind und es sich um das richtige Modell und die richtige Version handelt, die bestellt wurde.
- Überprüfung auf Schäden: Es wird eine Sichtprüfung durchgeführt, um sicherzustellen, dass während des Versands und der Handhabung keine Schäden aufgetreten sind.
Notiz: In den meisten Branchen, die die IQ OQ PQ-Methode anwenden, sollte die Ausrüstung mit Konformitätszertifikaten (CE-, FDA-Kennzeichnungen usw.), ggf. weiteren Zertifikaten und möglicherweise herstellereigenen Testergebnissen geliefert werden. Diese Dokumente sind genauso wichtig wie die physischen Waren selbst. Tatsächlich sollte eine Lieferung abgelehnt werden, wenn diese Dokumente nicht beiliegen oder im Voraus (zumindest in der Quarantänezone) eingegangen sind.
Verwandter Tipp: Vertrauen Sie der Firmware, aber überprüfen Sie sie. Die Packliste ist das absolute Minimum. Konzentrieren Sie sich auf die kritischen Komponenten und vor allem auf die Software- und Firmware-Versionen. Ein Lieferant liefert möglicherweise eine neuere, „bessere“ Version, die für Ihren Prozess nicht validiert wurde. Machen Sie vor der Abfahrt des Lieferfahrers und vor der Unterzeichnung der Versanddokumente hochauflösende Fotos der Typenschilder aller kritischen Komponenten (Motoren, Pumpen, Steuerungen) und des Hauptgeräteschilds. Schalten Sie die Steuerungseinheit nach Möglichkeit nur ein, um die Firmware-/Software-Version auf dem Startbildschirm zu überprüfen. Vergleichen Sie diese mit der Version in Ihrer Benutzeranforderungsspezifikation (URS) oder Bestellung. Es ist viel einfacher, die Lieferung an der Tür abzulehnen, als sich später mit der Diskrepanz auseinanderzusetzen.
3. Installation und Verbindungsüberprüfung
Dies ist der Kern des IQ-Prozesses, bei dem die physische Installation unter die Lupe genommen wird:

- Richtige Montage und Platzierung: Überprüfen, ob die Ausrüstung gemäß den Anweisungen und technischen Zeichnungen des Herstellers zusammengebaut und positioniert ist.
- Versorgungsanschlüsse: Dies ist ein kritischer Schritt, bei dem alle Verbindungen zu wichtigen Diensten bestätigt werden. Dazu gehört die Überprüfung, dass:
- Elektrisch: die Stromversorgung der erforderlichen Spannung und Phase entspricht und dass die richtigen Erdungs- und Sicherheitsschaltkreise vorhanden sind.
- Sanitär: Anschlüsse für Wasser, Dampf oder Abwasser sind fachgerecht installiert, dicht und aus den entsprechenden Materialien gefertigt.
- Gase und Druckluft: alle pneumatischen und gasförmigen Leitungen korrekt angeschlossen sind und Druck und Qualität den Spezifikationen entsprechen.
- Zusatzausrüstung: Stellen Sie sicher, dass alle unterstützenden oder peripheren Geräte ebenfalls korrekt installiert und angeschlossen sind.
- Softwareinstallation:bei computergesteuerten Systemen die Überprüfung der korrekten Installation der Software, der Kommunikation mit dem...
You have read 14% of the article. The rest is for our community. Already a member? Einloggen
(und auch um unsere Originalinhalte vor Scraping-Bots zu schützen)
Innovation.world Gemeinschaft
Anmelden oder Registrieren (100% kostenlos)
Lesen Sie den Rest dieses Artikels und alle Inhalte und Tools, die nur für Mitglieder zugänglich sind.
Nur echte Ingenieure, Hersteller, Designer und Marketingfachleute.
Kein Bot, kein Hater, kein Spammer.
Verwandte Lesungen
- Validierungsmasterplan (VMP): das strategische Dokument auf hoher Ebene, das die allgemeine Validierungsphilosophie, den Umfang, die Verantwortlichkeiten und die spezifischen zu validierenden Systeme und Prozesse des Unternehmens definiert.
- Benutzeranforderungsspezifikation (URS): Das grundlegende Dokument beschreibt detailliert, was die Ausrüstung oder der Prozess aus Sicht des Endbenutzers und der Qualität leisten soll und bildet die Basis für alle nachfolgenden Tests.
- Werksabnahmeprüfung (FAT) und Standortabnahmeprüfung (SAT): Von Ingenieuren geleitete Vortests, die häufig zusammen mit dem Anbieter durchgeführt werden, um die Funktionalität der Geräte vor dem Versand (FAT) und nach der Installation (SAT) zu überprüfen. Dies kann zur Optimierung von IQ/OQ genutzt werden.
- Computersystemvalidierung (CSV): eine spezialisierte Validierungsdisziplin, die parallel zu IQ OQ PQ läuft und sich auf die Integrität, Sicherheit und Zuverlässigkeit der Software und der Computersysteme konzentriert, die die Geräte steuern.
- Konformität mit 21 CFR Part 11: die spezifischen FDA-Vorschriften für elektronische Aufzeichnungen und elektronische Signaturen, eine kritische Komponente von CSV zur Gewährleistung der Datenintegrität, Prüfpfade und Zugriffskontrolle.
- Reinigungsvalidierung: Ein manchmal separater, aber verwandter Validierungsprozess, der an derselben Ausrüstung durchgeführt wird, um nachzuweisen, dass ein Reinigungsverfahren Produktrückstände, Reinigungsmittel und mikrobielle Verunreinigungen wirksam und konsistent entfernen kann (wir empfehlen dringend, ihn in das IQ OQ PQ-Protokoll aufzunehmen, da der Reinigungsstatus ein integraler Bestandteil der Produkt- und Prozessleistung ist).
- Messsystemanalyse (MSA) / Gage R&R: die statistische Methodik, die zur Validierung der *Testmethoden* selbst verwendet wird, um sicherzustellen, dass das zur Beurteilung der Produktqualität verwendete Messsystem genau, präzise, wiederholbar und reproduzierbar ist.
- Risikobasierte Validierung (mittels FMEA/HACCP): die Methodik zur Identifizierung potenzieller Fehlermodi und Risiken für die Produktqualität, sodass die Validierungsbemühungen auf die kritischsten Prozessparameter (CPPs) konzentriert werden können.
- Kritische Qualitätsattribute (CQAs) und kritische Prozessparameter (CPPs): die spezifischen, definierten Produktmerkmale (CQAs) und Prozessvariablen (CPPs), die kontrolliert werden müssen, um die gewünschte Produktqualität sicherzustellen, die die Akzeptanzkriterien für PQ bilden.
- Prozess-Fähigkeit Analyse (Cpk/Ppk): die statistische Analyse, die auf PQ-Daten durchgeführt wird, um zu quantifizieren, wie gut ein Prozess innerhalb seiner Spezifikationsgrenzen zentriert ist und wie viel Variabilität er aufweist, wodurch eine numerische Bewertung seiner „Fähigkeit“ bereitgestellt wird.
- Kontinuierliche Prozessüberprüfung (CPV): die moderne „Stufe 3“ der Prozessvalidierung, die die laufende Überwachung der Prozessparameter und Qualitätsmerkmale während der Routineproduktion umfasst, um sicherzustellen, dass der Prozess in einem konstanten Kontrollzustand bleibt.
- Änderungskontrollmanagement: das formelle Qualitätssystemverfahren zur Bewertung, Dokumentation und Genehmigung aller vorgeschlagenen Änderungen an einem validierten System oder Prozess, um sicherzustellen, dass dieser nicht unbeabsichtigt seinen validierten Zustand verlässt.
- Abweichungs- und CAPA-Management: das System zur Untersuchung, Bestimmung der Grundursache und Umsetzung von Korrektur- und Vorbeugemaßnahmen für alle unerwarteten Veranstaltungen oder Fehler, die während oder nach der Validierung auftreten.
- Retrospektive Validierung: Ein veralteter und heute kaum noch akzeptabler Validierungsansatz, der auf der Analyse historischer Produktionsdaten eines bestehenden, nicht validierten Prozesses basiert, um nachzuweisen, dass dieser unter Kontrolle gelaufen ist.
- Validierungszusammenfassungsbericht (VSR): das abschließende, schlüssige Dokument, das den gesamten Validierungsaufwand (IQ OQ PQ) zusammenfasst, die wichtigsten Ergebnisse präsentiert, auf etwaige Abweichungen eingeht und das System oder den Prozess offiziell als validiert und für die kommerzielle Nutzung geeignet erklärt.
Externe Links zur IQ OQ PQ Prozessvalidierung
Internationale Standards
(Bewegen Sie den Mauszeiger über den Link, um unsere Inhaltsbeschreibung anzuzeigen)
Glossar der verwendeten Begriffe
American Society for Testing and Materials (ASTM): eine internationale Normungsorganisation, die freiwillige, auf Konsens beruhende technische Normen für Materialien, Produkte, Systeme und Dienstleistungen entwickelt und veröffentlicht, mit dem Ziel, Qualität und Sicherheit in verschiedenen Branchen zu verbessern.
Calculation of Process Capability (Cpk): Ein statistisches Maß, das die Fähigkeit eines Prozesses bewertet, Ergebnisse innerhalb festgelegter Grenzen zu erzielen. Die Berechnung erfolgt durch die Bewertung des Abstands zwischen dem Prozessmittelwert und der nächstgelegenen Spezifikationsgrenze, normalisiert durch die Prozessstandardabweichung.
Computer Numerically Controlled (CNC): Ein Herstellungsprozess, bei dem programmierte Computersoftware zur Steuerung von Werkzeugmaschinen verwendet wird, um einen präzisen und automatisierten Betrieb für Aufgaben wie Schneiden, Fräsen, Bohren und Gravieren von Materialien zu ermöglichen.
Continuous Integration/Continuous Deployment (CI/CD): Eine Softwareentwicklungspraxis, die die Integration von Codeänderungen und die Bereitstellung in Produktionsumgebungen automatisiert und so durch automatisierte Tests und Überwachung häufige Updates, schnellere Bereitstellung und eine verbesserte Zusammenarbeit zwischen Entwicklungs- und Betriebsteams ermöglicht.
Corrective Action and Preventative Action (CAPA): Ein systematischer Ansatz zur Identifizierung, Untersuchung und Behebung von Nichtkonformitäten und potenziellen Problemen, um ein erneutes Auftreten zu verhindern und die Einhaltung gesetzlicher Standards in Qualitätsmanagementsystemen sicherzustellen.
Critical Control Points (CCP): Spezifische Phasen eines Prozesses, in denen Kontrollen angewendet werden können, um Gefahren für die Lebensmittelsicherheit zu verhindern, zu beseitigen oder auf ein akzeptables Maß zu reduzieren. Die Identifizierung dieser Punkte ist für eine effektive Gefahrenanalyse und ein kritisches Kontrollmanagement in Lebensmittelproduktionssystemen von entscheidender Bedeutung.
current Good Manufacturing Practice (cGMP): Ein System, das sicherstellt, dass Produkte konsistent gemäß Qualitätsstandards hergestellt und kontrolliert werden. Es umfasst Vorschriften und Richtlinien für Herstellungsprozesse, Einrichtungen, Ausrüstung und Personal, um Sicherheit, Qualität und Wirksamkeit in der Pharma- und Lebensmittelindustrie sowie in anderen regulierten Branchen zu gewährleisten.
Defects Per Million Opportunities (DPMO): Eine in der Qualitätskontrolle verwendete Messung, die die Anzahl der Fehler in einem Prozess pro einer Million Fehlermöglichkeiten quantifiziert. Die Berechnung erfolgt durch Division der Anzahl der Fehler durch die Gesamtzahl der Möglichkeiten und Multiplikation mit einer Million.
Define Measure Analyze Improve Control (DMAIC): Eine datengesteuerte Qualitätsstrategie, die in Six Sigma zur Prozessverbesserung verwendet wird und aus fünf Phasen besteht: Identifizierung des Problems, Messung der aktuellen Leistung, Analyse der Daten zur Identifizierung der Ursachen, Verbesserung der Prozesse auf Grundlage der Erkenntnisse und Kontrolle der zukünftigen Leistung zur Aufrechterhaltung der Verbesserungen.
Design Validation (DV): Ein Prozess, der durch Tests und Bewertungen sicherstellt, dass ein Produkt die angegebenen Anforderungen und den vorgesehenen Verwendungszweck erfüllt. Dabei wird bestätigt, dass das Design seinen Zweck erfüllt und unter realen Bedingungen ordnungsgemäß funktioniert.
Failure Mode and Effects Analysis (FMEA): Eine systematische Methode zur Bewertung potenzieller Fehlermodi innerhalb eines Systems, Prozesses oder Produkts, zur Beurteilung ihrer Auswirkungen auf die Leistung und zur Priorisierung von Risiken, um Zuverlässigkeit und Sicherheit durch Korrekturmaßnahmen zu verbessern.
Food and Drug Administration (FDA): eine Bundesbehörde des US-Gesundheitsministeriums, die für die Regulierung der Lebensmittelsicherheit, Arzneimittel, Medizinprodukte, Kosmetika und Tabakprodukte zuständig ist, um durch wissenschaftliche Bewertung und Durchsetzung von Konformitätsstandards die öffentliche Gesundheit und Sicherheit zu gewährleisten.
Good Manufacturing Practice (GMP): Ein System, das die konsistente Herstellung und Kontrolle von Produkten gemäß Qualitätsstandards gewährleistet und so die Risiken in der Pharmaproduktion und verwandten Branchen minimiert. Es umfasst Richtlinien für Herstellungsprozesse, Anlagenbedingungen, Personalqualifikationen und Dokumentationspraktiken, um die Sicherheit und Wirksamkeit der Produkte zu gewährleisten.
Hazard Analysis and Critical Control Points (HACCP): Ein systematischer Ansatz zur Lebensmittelsicherheit, der Gefahren an kritischen Punkten des Produktionsprozesses identifiziert, bewertet und kontrolliert, um durch Lebensmittel verursachte Krankheiten zu verhindern und die Produktsicherheit zu gewährleisten.
Installation Qualification (IQ): Ein dokumentierter Prozess zur Überprüfung, ob Geräte oder Systeme gemäß den Spezifikationen installiert werden, einschließlich der Bewertung von Versorgungseinrichtungen, Umgebungsbedingungen und der Einhaltung von Designanforderungen, um die Bereitschaft zur Betriebsqualifizierung sicherzustellen.
Measurement System Analysis (MSA): Eine statistische Methode zur Bewertung der Genauigkeit, Präzision und Zuverlässigkeit von Messvorgängen und -instrumenten. Sie stellt sicher, dass die gesammelten Daten für die Entscheidungsfindung bei der Qualitätskontrolle und Prozessverbesserung gültig und konsistent sind.
Operational Qualification (OQ): Ein Validierungsprozess, der sicherstellt, dass Geräte oder Systeme innerhalb definierter Grenzen gemäß den angegebenen Anforderungen funktionieren und bestätigt, dass sie in ihrer Betriebsumgebung die vorgesehene Leistung erbringen.
Performance Qualification (PQ): Ein Prozess, der überprüft, ob ein System oder Gerät unter realen Bedingungen gemäß den angegebenen Anforderungen funktioniert und sicherstellt, dass es seine beabsichtigte Funktion innerhalb vorgegebener Grenzen durchgängig erfüllt.
Process Capability Index (Cpk): Ein statistisches Maß, das quantifiziert, wie gut ein Prozess innerhalb festgelegter Grenzen Ergebnisse erzielen kann. Es gibt die Beziehung zwischen dem Prozessmittelwert und der nächstgelegenen Spezifikationsgrenze an, angepasst an die Prozessvariabilität.
Process Performance Index (Ppk): Ein statistisches Maß, das quantifiziert, wie gut ein Prozess Spezifikationsgrenzen einhält. Die Berechnung erfolgt anhand des Prozessmittelwerts und der Standardabweichung. Es zeigt die Fähigkeit eines Prozesses an, Ergebnisse innerhalb definierter Grenzen zu erzielen, wobei sowohl Variabilität als auch Zentrierung berücksichtigt werden.
Product Lifecycle Management (PLM): Ein systematischer Ansatz zur Verwaltung des Lebenszyklus eines Produkts von der Entstehung über die Konstruktion und Fertigung bis hin zu Service und Entsorgung. Dabei werden Menschen, Prozesse, Daten und Technologie integriert, um die Produktqualität zu verbessern, die Markteinführungszeit zu verkürzen und die Zusammenarbeit zwischen den Beteiligten zu verbessern.
Production Part Approval Process (PPAP): Ein standardisiertes Verfahren, das in der Fertigung verwendet wird, um sicherzustellen, dass Lieferanten die Qualitätsanforderungen vor der Massenproduktion erfüllen. Dazu gehört die Dokumentation und Validierung von Designspezifikationen, Prozessfähigkeiten und Produktionsmustern, um die Einhaltung der Kundenerwartungen zu bestätigen.
Quality Management System (QMS): ein strukturiertes System aus Prozessen, Verfahren und Verantwortlichkeiten, das darauf abzielt, eine gleichbleibende Qualität der Produkte und Dienstleistungen sicherzustellen, eine kontinuierliche Verbesserung zu ermöglichen und die Anforderungen der Kunden und der Behörden zu erfüllen.
Standard Operating Procedure (SOP): Eine Reihe schrittweiser Anweisungen, die den Mitarbeitern dabei helfen sollen, Routinevorgänge konsistent und effizient durchzuführen und so die Einhaltung von Vorschriften und Qualitätsstandards sicherzustellen.
Statistical Process Control (SPC): Eine Methode der Qualitätskontrolle, bei der statistische Techniken zum Überwachen und Steuern eines Prozesses eingesetzt werden. Durch die Identifizierung von Abweichungen und die Aufrechterhaltung einer konsistenten Ausgabe innerhalb festgelegter Grenzen wird sichergestellt, dass dieser sein volles Potenzial ausschöpft.
User Requirement Specification (URS): Ein Dokument, das die Bedürfnisse und Erwartungen der Benutzer an ein System oder Produkt detailliert beschreibt und funktionale und nicht-funktionale Anforderungen umreißt, um die Entwicklung zu leiten und die Ausrichtung an den Benutzerzielen sicherzustellen.























Verwandte Artikel
Kontaminationskontrollstrategie und Best Practices für Reinräume 26
Von GMP zu cGMP: Der vollständige Mastering-Leitfaden
Die Strategien „Lone Nut“, „First Follower“ und „Fast Follower“
Die 20 besten Verwendungsmöglichkeiten von Proxies im Engineering
Wie man Eis an Eskimos Verkauft (oder Marketing-Spielereien)
Greenwashing: Die 15 besten Tipps eines Gentlemans zur exquisiten Täuschung