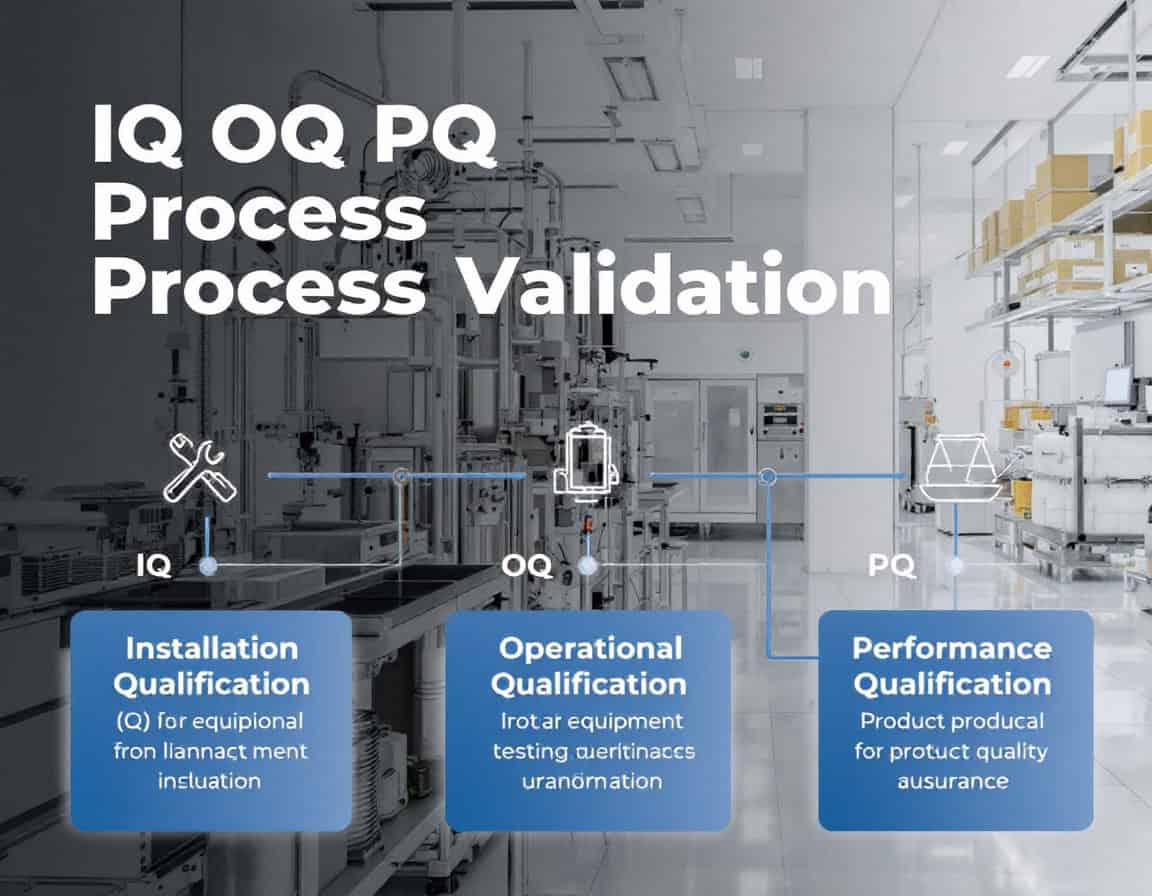
IQ OQ PQ 流程 验证 方法论,即安装确认 (IQ)、运行确认 (OQ) 和性能确认 (PQ) 顺序,构成了既定的流程。 框架 for process validation, serving as the systematic generation of documented evidence that a 制造业 process is in a state of control. This methodology follows a deliberate and logical progression, beginning with the static, documented 确认 设备安装正确(IQ),动态确认设备在其规定的范围内可靠运行(OQ),最终证明集成过程能够持续生产出符合所有质量属性的产品(PQ)。
整个序列旨在提供客观、可追溯的证据,证明该过程在商业使用之前是稳健的、可重复的并且适合其预期用途。
这种方法是 cGMP 管理的行业(如制药、生物制剂和 医疗器械 制造工艺的一致性与患者安全直接相关。其应用并非用于初步研究,而是在工艺开发完成后、获准进行常规商业化生产之前进行的关键验证阶段。
关键要点

- 验证的输出是证据,而不是产品。
- “最坏情况” OQ 测试涉及组合压力源。
- PQ 验证的是集成过程,而不仅仅是设备。
- “三批”规则体现了可重复性。
- PQ 的成功取决于一致性,而不仅仅是通过规范。
- 该协议是一种合同;偏差必须有合理的依据并有充分的记录。
- 利用供应商文档(FAT/SAT),但不要取代您自己的 OQ
- 验证定义了生命周期的开始,而不是项目的结束。
- 任何“同类”的改变都需要理由。
- 人员培训是OQ和PQ的先决条件
安装确认(IQ)
安装确认 (IQ) 是 IQ OQ PQ 流程验证方法的初始和基础阶段。它是一个正式的、记录在案的流程,用于验证并确认设备、系统和辅助组件已完全按照制造商的建议和用户的设计规范进行交付、安装和配置。
IQ 的核心原则是提供文件证据证明安装正确,并为后续的鉴定阶段创造合适、安全的环境。
IQ 的目的和重要性: 设备质量验证 (IQ) 的主要目标是建立设备验证的基准。在开始任何操作测试之前,至关重要的是确保设备实际存在、正确组装并放置在合适的环境中。此阶段可以降低与不当安装相关的风险,否则可能导致设备故障、产品质量问题和安全隐患。
安装确认的关键阶段和活动
IQ 流程是系统的,涉及几个详细的阶段,通常通过预先批准的协议和清单进行管理。
1. 安装前和规划
在设备到达或订购之前,质量评估流程就已从周密的规划开始。这包括:
- 场地准备情况验证: 确保指定位置已为新设备做好准备。这包括检查是否有足够的占地面积、结构支撑,以及在正确的连接点是否配备必要的公用设施。
- 环境条件: 验证安装环境是否符合制造商对温度、湿度和清洁度的规范。
- 收集文件: 收集所有必要的文件,例如采购订单、制造商手册、设计规范、工程图和任何综合测量仪器的校准证书。
提示: 走一遍路线,并建模体积。不要只看二维平面图。最常见且代价高昂的IQ失败往往发生在这里。与关键利益相关者(设施部门、工程部门和供应商的项目经理)一起,沿着从装卸码头到最终安装点的规划路线走一遍。使用一个简单的木制或PVC框架,根据机器的最大高度、宽度和长度尺寸建造。沿着整个路径实际搬运这个“幽灵”框架。这将揭示图纸经常遗漏的诸如门道、低垂管道、狭窄拐角和电梯容量等净空问题。 此外,不仅要验证最终位置的地板承载能力,还要验证整个运输路径的地板承载能力。

2. 设备接收与验证
设备交付后,将进行彻底检查:
- 组件验证: 根据装箱单和采购订单对交付的物品进行仔细检查,以确认所有组件,包括 软件已收到,型号和版本与订购的一致。
- 损坏检查: 进行目视检查以确保在运输和搬运过程中没有发生损坏。
笔记: 在大多数采用IQ OQ PQ方法的行业中,设备应附带符合性证书(例如CE、FDA标志等),以及其他证书,以及一些制造商自己的测试结果。这些文件应与实物本身同等重要。事实上,如果未包含这些文件或未提前收到(至少在隔离区),则应拒绝收货。
相关提示: 信任,但要验证固件。装箱单只是最低限度的清单。您真正应该关注的是关键组件,以及最重要的软件和固件版本。供应商可能会发送一个更新、“更好”的版本,而该版本尚未经过您的流程验证。在送货司机离开之前以及在您签署任何装运单据之前,请拍摄所有关键组件(电机、泵、控制器)铭牌和主设备铭牌的高分辨率照片。如果可能,仅在启动屏幕上验证固件/软件版本时才打开控制器单元。将此版本与您的用户需求规范 (URS) 或采购订单中指定的版本进行交叉比对。当场拒收货物远比事后处理差异容易得多。
3. 安装和连接验证
这是 IQ 流程的核心,其中对物理安装进行了仔细检查:

- 正确的组装和放置: 验证设备是否按照制造商的说明和工程图进行组装和定位。
- 公用设施连接: 这是至关重要的一步,涉及确认所有基本服务的连接。这包括检查:
- 电气: 电源是否与所需电压和相位相匹配,并且接地和安全电路是否正确到位。
- 管道: 水、蒸汽或排水连接件安装正确、无泄漏且采用合适的材料制成。
- 气体和压缩空气: 所有气动和燃气管路均已正确连接,并且 压力 质量符合规格要求。
- 辅助设备: 确保任何支持或外围设备也正确安装和连接。
- 软件安装: 对于计算机控制系统来说,这包括核查软件是否安装正确、, 沟通 硬件已安装完毕,访问控制也已到位。
提示: 实时红线标注图纸。切勿想当然地认为 P&ID(管道和仪表图)或电气图纸 100% 准确地代表最终安装。供应商的安装人员总会在现场进行细微的实际调整。请指派一位您自己的工程师或合格技术人员,带着一套正式图纸和一支红笔跟随供应商的安装人员。当安装人员进行连接时,您的工程师应实际描摹每条线路(管道、导管、电缆),并将任何与图纸的偏差直接标记在图纸上。这份“竣工”红线标注的图纸是一份宝贵的文档。在 IQ 结束时,您可以将这份红线标注的版本扫描为真实安装的正式记录,这对于未来的故障排除、维护和审计至关重要。
4. 文件和记录保存

在整个质量审查过程中,细致的记录至关重要。这包括:
- 记录序列号: 记录主要设备和所有关键部件的序列号和型号。
- 编译文档包: 创建一个活页夹或电子文件夹,其中包含所有相关文件,例如手册、图纸、校准记录和已完成的 IQ 协议。
- 制定维护和校准计划: 按照制造商的建议制定并记录日常维护和校准的计划。
提示: 你的主要目标是在你的设备中建立永久的活页夹或数字记录 产品生命周期 管理 (PLM) 或质量管理系统 (QMS),使其成为唯一真实来源。在质量评估 (IQ) 开始之前,为系统中的设备创建唯一的资产记录。每份文档生成后(从供应商证书到签名的协议页),立即扫描并上传,并将其直接链接到此中央资产记录。这种做法超越了简单的归档,创建了一个可控且权威的 数字孪生 从一开始就对设备文档进行管理。使用系统的电子签名工作流程,将最终的 IQ 包提交审核和批准,从而提供安全合规的审计跟踪。更重要的是,通过将 IQ 文档链接到其他企业系统,构建关系模型。
5. 最终 IQ 报告及批准

对于这类行业来说,这很简单:完成所有检查后,将生成最终的 IQ 报告。该报告总结了已执行的活动,提供了记录的证据,详细说明了发现的任何偏差或差异及其解决方法,并得出结论,确定设备是否通过了安装确认。然后,该报告将由相关人员(例如质量保证部门)审核并正式批准,正式结束 IQ 阶段,并允许启动运行确认 (OQ)。
提示: 将偏差视为系统稳健的证据,而非故障。IQ 的目标并非追求零偏差的“完美”运行,而是真实记录安装后的状态。管理良好的偏差是健康透明的质量体系的标志。发现差异时,应立即将其记录为 IQ 协议中的计划偏差。然后,进行正式的风险评估。评估是在 OQ 开始前制定更换部件的纠正措施,还是采取其他方法?在最终 IQ 报告中记录整个过程,可以向审核员表明您的系统一丝不苟、基于风险且处于可控状态。
The rest of this article is reserved for members
To limit scraping bots (currently 40,000 hits per day!),
we had to restrict access to full articles and tools to registered members only.
to access all the rest.