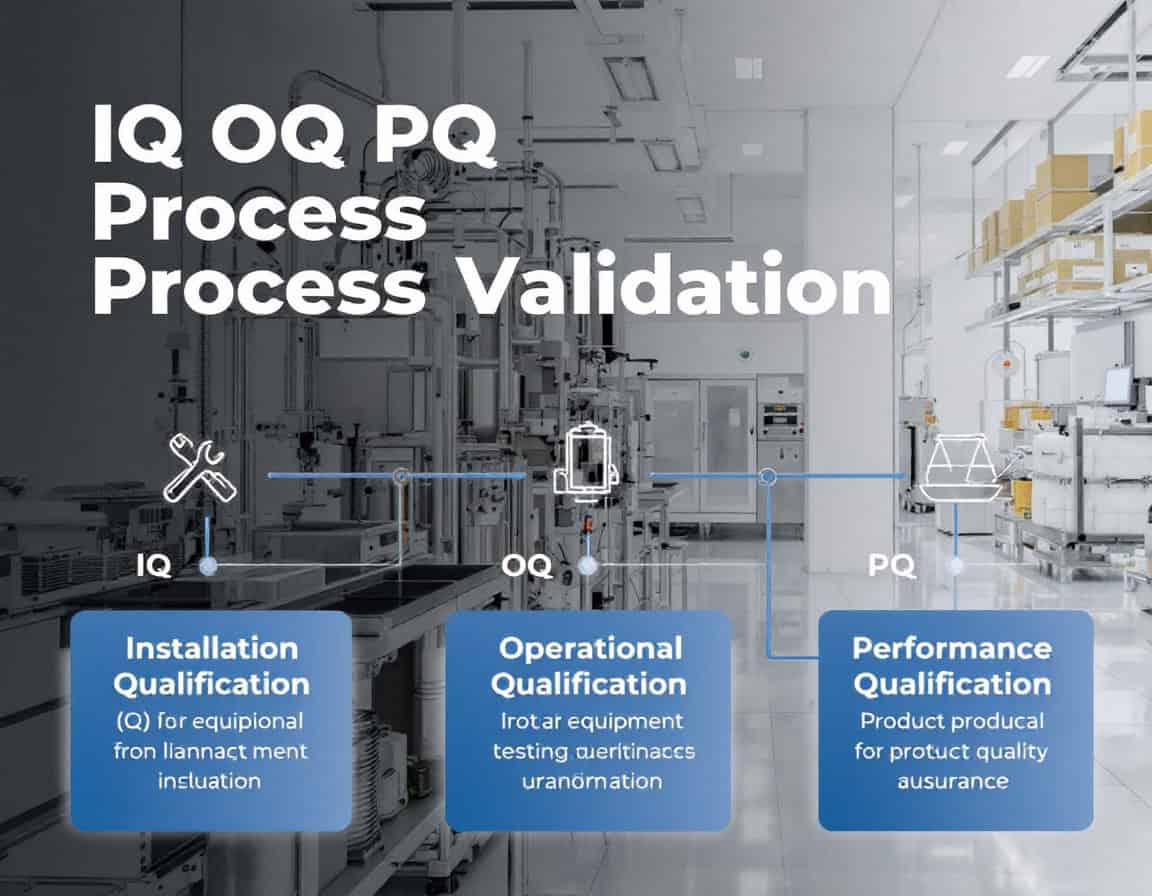
Le processus IQ OQ PQ validation La méthodologie, qui désigne la séquence de qualification d'installation (QI), de qualification opérationnelle (QO) et de qualification de performance (QP), constitue la méthode établie cadre pour la validation des procédés, servant à générer systématiquement des preuves documentées qu'un procédé de fabrication est maîtrisé. Cette méthodologie suit une progression délibérée et logique, commençant par l'analyse statique et documentée. vérification que l'équipement est correctement installé (IQ), en passant à la confirmation dynamique que l'équipement fonctionne de manière fiable sur toutes ses plages spécifiées (OQ), et en aboutissant à la démonstration que le processus intégré produit de manière constante un produit répondant à tous les attributs de qualité (PQ).
L’ensemble de la séquence est conçu pour fournir une preuve objective et traçable que le processus est robuste, reproductible et adapté à l’usage prévu avant toute utilisation commerciale.
Cette méthodologie est une condition réglementaire non négociable dans les industries régies par les BPF, telles que les produits pharmaceutiques, biologiques et dispositif médical fabricationDans ce contexte, la constance du processus est directement liée à la sécurité des patients. Son application ne concerne pas la recherche initiale, mais constitue l'étape critique de validation menée après la finalisation du développement du processus et avant son approbation pour la production commerciale de routine.
A Retenir

- Le résultat de la validation est une preuve, pas un produit.
- Les tests OQ « du pire des cas » impliquent des facteurs de stress combinés.
- PQ valide le processus intégré, pas seulement l’équipement.
- La règle des « trois lots » démontre la reproductibilité.
- Le succès du PQ est défini par la cohérence, et pas seulement par la réussite des spécifications.
- Le protocole est un contrat ; les écarts doivent être justifiés et bien documentés.
- Tirez parti de la documentation du fournisseur (FAT/SAT), mais ne remplacez pas votre propre OQ
- La validation définit le début du cycle de vie, et non la fin d’un projet.
- Tout changement « à l’identique » nécessite une justification.
- La formation du personnel est une condition préalable à la QO et à la QP
Qualification d'installation (QI)
La Qualification d'Installation (QI) est la phase initiale et fondamentale de la méthodologie de validation du processus QI/QO/QP. Il s'agit d'un processus formel et documenté qui vérifie et confirme que les équipements, systèmes et composants auxiliaires ont été livrés, installés et configurés conformément aux recommandations du fabricant et aux spécifications de conception de l'utilisateur.
Le principe fondamental d’IQ est de fournir des preuves documentées que l’installation est correcte et crée un environnement approprié et sûr pour les phases de qualification ultérieures.
Le but et l’importance du QI : L'objectif principal de la QI est d'établir une base de référence pour la validation de l'équipement. Avant tout essai opérationnel, il est crucial de s'assurer que l'équipement est physiquement présent, correctement assemblé et situé dans un environnement approprié. Cette phase permet d'atténuer les risques liés à une installation incorrecte, susceptible d'entraîner un dysfonctionnement de l'équipement, des problèmes de qualité du produit et des risques pour la sécurité.
Étapes et activités clés de la qualification d'installation
Le processus IQ est systématique et implique plusieurs étapes détaillées, généralement gérées par un protocole et une liste de contrôle pré-approuvés.
1. Pré-installation et planification

Avant même l'arrivée ou la commande de l'équipement, le processus de QI commence par une planification minutieuse. Cela comprend :
- Vérification de l'état de préparation du site : S'assurer que l'emplacement prévu est prêt à accueillir le nouvel équipement. Cela implique de vérifier l'espace au sol, le support structurel et la disponibilité des services publics nécessaires aux points de raccordement appropriés.
- Conditions environnementales : vérifier que l'environnement d'installation répond aux spécifications du fabricant en matière de température, d'humidité et de propreté.
- Rassembler la documentation : rassembler tous les documents essentiels, tels que le bon de commande, les manuels du fabricant, les spécifications de conception, les dessins techniques et les certificats d'étalonnage pour tous les instruments de mesure intégrés.
Conseil: Parcourez le parcours et modélisez le volume. Ne vous contentez pas de consulter un plan d'étage en 2D. C'est là que se produisent les pannes d'intelligence les plus courantes et les plus coûteuses. Parcourez le parcours prévu, du quai de chargement au point d'installation final, avec vos principaux intervenants (services techniques, ingénierie et chef de projet du fournisseur). Utilisez un simple cadre en bois ou en PVC, construit aux dimensions maximales de hauteur, de largeur et de longueur de la machine. Transportez physiquement ce cadre « fantôme » tout au long du parcours. Cela révélera les problèmes de dégagement liés aux portes, aux tuyaux bas, aux angles étroits et aux capacités des ascenseurs, souvent ignorés par les plans. Vérifiez également la capacité de charge du sol non seulement à l’emplacement final, mais tout au long du trajet de transit.

2. Réception et vérification de l'équipement
Une fois le matériel livré, une inspection approfondie est effectuée :
- Vérification des composants : Les articles livrés sont minutieusement vérifiés par rapport au bon de livraison et au bon de commande afin de confirmer que tous les composants, y compris logiciel, ont été reçues et correspondent au modèle et à la version commandés.
- Inspection des dommages : une inspection visuelle est effectuée pour s'assurer qu'aucun dommage n'est survenu pendant l'expédition et la manutention.
Note: Dans la plupart des industries appliquant la méthodologie QI/QO/QP, l'équipement doit être accompagné de ses certificats de conformité (marquages CE/FDA…), éventuellement d'autres certificats et, éventuellement, de résultats de tests internes au fabricant. Ces documents doivent être considérés au même titre que les biens eux-mêmes. En effet, une livraison doit être refusée si ces documents ne sont pas inclus ou reçus à l'avance (au moins en zone de quarantaine).
Conseil connexe : Faites confiance, mais vérifiez le micrologiciel. La liste de colisage est le strict minimum. Concentrez-vous sur les composants critiques et, surtout, sur les versions logicielles et micrologicielles. Un fournisseur pourrait vous livrer une version plus récente et « améliorée », mais non validée pour votre processus. Avant le départ du livreur et avant de signer les documents d'expédition, prenez des photos haute résolution des plaques signalétiques de tous les composants critiques (moteurs, pompes, contrôleurs) et de la plaque signalétique principale. Si possible, allumez le contrôleur uniquement pour vérifier la version du micrologiciel/logiciel sur l'écran de démarrage. Comparez-la à la version spécifiée dans votre cahier des charges utilisateur (URS) ou votre bon de commande. Il est bien plus facile de refuser la livraison à la porte que de gérer l'écart ultérieurement.
3. Vérification de l'installation et de la connexion
C'est le cœur du processus IQ, où l'installation physique est examinée :

- Montage et placement corrects : vérifier que l'équipement est assemblé et positionné conformément aux instructions du fabricant et aux dessins techniques.
- Raccordements aux services publics : Il s'agit d'une étape cruciale qui implique de confirmer toutes les connexions aux services essentiels. Cela comprend la vérification des points suivants :
- Électrique: l'alimentation électrique correspond à la tension et à la phase requises, et que des circuits de mise à la terre et de sécurité appropriés sont en place.
- Plomberie: les raccordements pour l'eau, la vapeur ou le drainage sont correctement installés, sans fuite et fabriqués avec les matériaux appropriés.
- Gaz et air comprimé : Toutes les conduites pneumatiques et de gaz sont correctement raccordées, et pression et la qualité est conforme aux spécifications.
- Équipements auxiliaires : s'assurer que tout équipement de support ou périphérique est également correctement installé et connecté.
- Installation du logiciel : Pour les systèmes informatisés, cela implique de vérifier que le logiciel est correctement installé. communication le matériel est installé et les contrôles d'accès sont en place.
Conseil: Relevez les plans en temps réel. Ne présumez jamais que le schéma P&ID (tuyauterie et instrumentation) ou les plans électriques sont des représentations exactes à 100 % de l'installation finale. L'installateur du fournisseur effectuera toujours des ajustements mineurs et pratiques sur site. Désignez l'un de vos ingénieurs ou techniciens qualifiés pour suivre l'installateur du fournisseur avec un jeu de plans officiels et un stylo rouge. Au fur et à mesure que l'installateur effectue les raccordements, votre ingénieur doit tracer physiquement chaque ligne (tuyau, conduit, câble) et signaler tout écart par rapport au plan directement sur l'impression. Ce plan « tel que construit » rehaussé de rouge est un document précieux. À la fin de l'IQ, vous numérisez cette version rehaussée de rouge comme enregistrement officiel de l'installation réelle, précieux pour les dépannages, la maintenance et les audits ultérieurs.
4. Documentation et tenue de registres

Tout au long du processus d'IQ, une documentation rigoureuse est primordiale. Cela comprend :
- Enregistrement des numéros de série : documenter les numéros de série et les numéros de modèle de l'équipement principal et de tous les composants critiques.
- Compilation d'un package de documentation : créer un classeur ou un dossier électronique contenant tous les documents pertinents, tels que les manuels, les dessins, les dossiers d'étalonnage et le protocole IQ complété.
- Établissement des calendriers de maintenance et d’étalonnage : établir et documenter les calendriers d'entretien de routine et d'étalonnage tels que recommandés par le fabricant.
Conseil: votre objectif principal est d'établir le classeur permanent ou l'enregistrement numérique de l'équipement dans votre Cycle de vie du produit Gestion du cycle de vie des produits (PLM) ou Système de gestion de la qualité (SMQ), ce qui en fait la source unique de référence. Avant le début de la QI, créez un enregistrement d'actif unique pour chaque équipement du système. Dès la génération de chaque document, des certificats fournisseurs aux pages de protocole signées, numérisez-le et téléchargez-le immédiatement, en le reliant directement à cet enregistrement d'actif central. Cette pratique va au-delà du simple archivage et crée un système contrôlé et fiable. jumeau numérique de la documentation de l'équipement dès sa création. Transférez le dossier IQ final pour examen et approbation grâce aux flux de signature électronique du système, qui offrent une piste d'audit sécurisée et conforme. Plus important encore, construisez un modèle relationnel en reliant la documentation IQ aux autres systèmes de l'entreprise.
5. Rapport final de QI et approbation

Rien de trop compliqué ici pour ce type d'industrie : une fois tous les contrôles terminés, un rapport final de QI est généré. Ce rapport résume les activités réalisées, présente les preuves documentées, détaille les écarts ou divergences constatés et leur résolution, et conclut si l'équipement a réussi la qualification d'installation. Ce rapport est ensuite examiné et approuvé formellement par le personnel concerné, notamment le service d'assurance qualité, concluant ainsi officiellement la phase de QI et autorisant le démarrage de la qualification opérationnelle (QO).
Conseil: Considérez les écarts comme la preuve d'un système robuste, et non comme une défaillance. L'objectif de la QI n'est pas d'obtenir un fonctionnement « parfait » sans écarts. Il s'agit de documenter fidèlement l'état d'installation. Un écart bien géré est le signe d'un système qualité sain et transparent. Lorsqu'un écart est détecté, documentez-le immédiatement comme un écart planifié dans le protocole de QI. Ensuite, menez une évaluation formelle des risques. Peut-il s'agir de mettre en place une action corrective pour remplacer la pièce avant le début de la QO ou d'adopter une approche différente ? La documentation de l'ensemble de ce processus dans le rapport final de QI montre aux auditeurs que votre système est méticuleux, axé sur les risques et maîtrisé.
The rest of this article is reserved for members
To limit scraping bots (currently 40,000 hits per day!),
we had to restrict access to full articles and tools to registered members only.
to access all the rest.