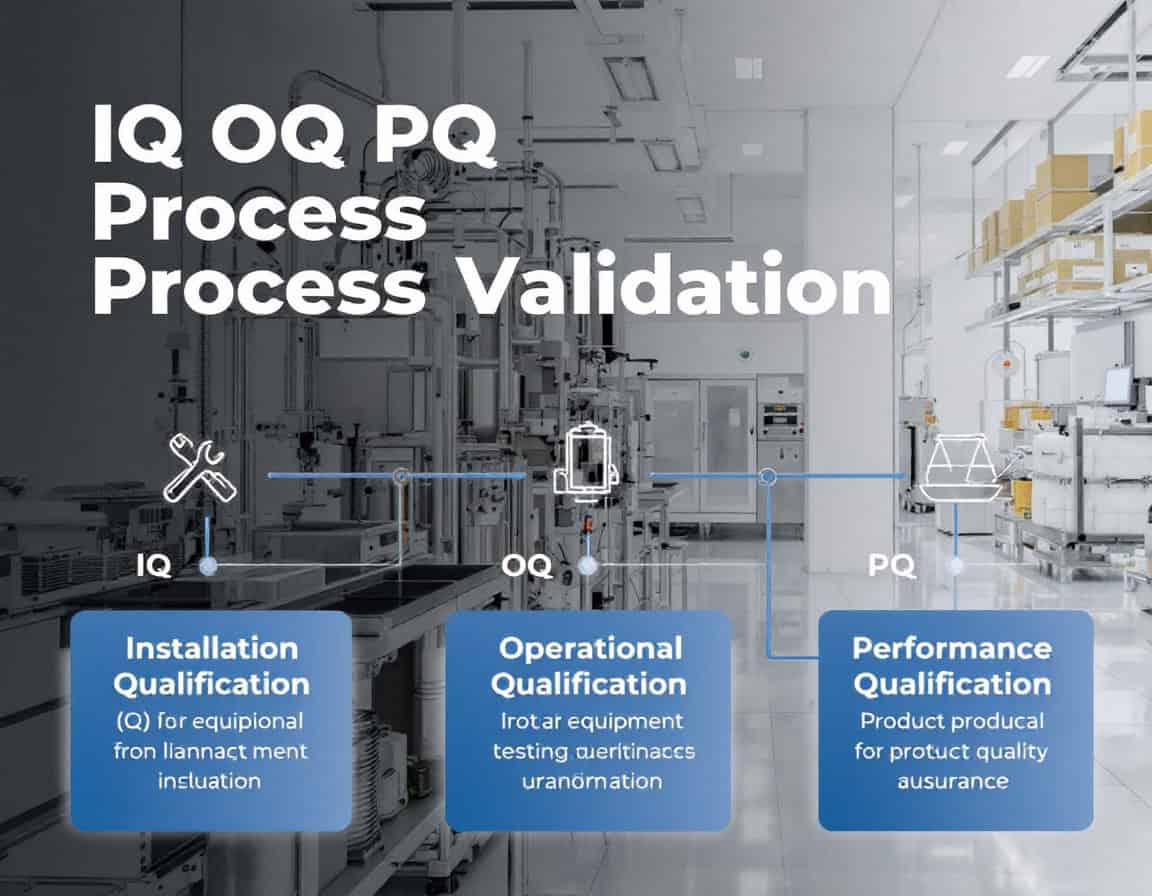
The IQ OQ PQ process validation methodology, standing for Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ) sequence constitutes the established framework for process validation, serving as the systematic generation of documented evidence that a manufacturing process is in a state of control. This methodology follows a deliberate and logical progression, beginning with the static, documented verification that equipment is installed correctly (IQ), moving to the dynamic confirmation that the equipment operates reliably across its specified ranges (OQ), and culminating in the demonstration that the integrated process consistently produces product meeting all quality attributes (PQ).
The entire sequence is designed to provide objective, traceable proof that the process is robust, reproducible, and fit for its intended purpose before commercial use.
This methodology is a non-negotiable regulatory prerequisite in industries governed by cGMP, such as pharmaceuticals, biologics, and medical device manufacturing, where process consistency is directly linked to patient safety. Its application is not for initial research but is the critical validation stage conducted after process development is finalized and before the process is approved for routine commercial production.
Key Takeaways

- Validation’s output is evidence, not product.
- “Worst-Case” OQ testing involves combined stressors.
- PQ validates the integrated process, not just the equipment.
- The “Three Batch” rule demonstrates reproducibility.
- PQ success is defined by consistency, not just passing spec.
- The protocol is a contract; deviations must be justified and well-documented.
- Leverage supplier documentation (FAT/SAT), but do not replace your own OQ
- Validation defines the start of the lifecycle, not the end of a project.
- Any “like-for-like” change requires justification.
- Personnel training is a prerequisite for OQ and PQ
Installation Qualification (IQ)
Installation Qualification (IQ) is the initial and foundational phase of the IQ OQ PQ process validation methodology. It is a formal, documented process that verifies and confirms that equipment, systems, and ancillary components have been delivered, installed, and configured in complete accordance with the manufacturer’s recommendations and the user’s design specifications.
The core principle of IQ is to provide documented evidence that the installation is correct and creates a suitable and safe environment for the subsequent qualification phases.
The purpose and importance of IQ: the primary goal of IQ is to establish a baseline for the equipment’s validation. Before any operational testing can begin, it is crucial to ensure that the equipment is physically present, correctly assembled, and situated in an appropriate environment. This phase mitigates risks associated with improper installation, which could otherwise lead to equipment malfunction, product quality issues, and safety hazards.
Key Stages and Activities in Installation Qualification
The IQ process is systematic and involves several detailed stages, typically managed through a pre-approved protocol and checklist.
1. Pre-Installation and Planning

Before the equipment even arrives, or is even ordered, the IQ process begins with careful planning. This includes:
- Site readiness verification: ensuring the designated location is prepared for the new equipment. This involves checking for adequate floor space, structural support, and the availability of necessary utilities at the correct connection points.
- Environmental conditions: verifying that the installation environment meets the manufacturer’s specifications for temperature, humidity, and cleanliness.
- Gathering documentation: collecting all essential documents, such as the purchase order, manufacturer’s manuals, design specifications, engineering drawings, and calibration certificates for any integrated measurement instruments.
Tip: walk the path and model the volume. Don’t just look at a 2D floor plan. The most common and costly IQ failures happen here. Literally walk the planned route from the loading dock to the final installation point with your key stakeholders (Facilities, Engineering, and the vendor’s project manager). Use a simple wooden or PVC frame built to the machine’s maximum height, width, and length dimensions. Physically carry this “ghost” frame along the entire path. This will reveal clearance issues with doorways, low-hanging pipes, tight corners, and elevator capacities that drawings often miss. Also, verify the floor loading capacity not just at the final spot, but along the entire transit path.

2. Equipment Receipt and Verification
Once the equipment is delivered, a thorough inspection is conducted:
- Component verification: the delivered items are meticulously checked against the packing list and purchase order to confirm that all components, including software, have been received and are the correct model and version ordered.
- Inspection for damage: a visual inspection is performed to ensure that no damage occurred during shipping and handling.
Note: in most industries practicing the IQ OQ PQ methodology, the equipment should come with its certificates of conformance (CE FDA markings …), eventually other certificates, and possibly some manufacturing own tests results. These documents should be considered in the same way and importance as the physical goods themselves. In fact a delivery should be refused if these documents are not included or received in advance (in quarantine zone at least).
Related tip: trust, but verify the Firmware. The packing list is the bare minimum. Your real focus should be on the critical components and, most importantly, the software and firmware versions. A vendor might ship a newer, “better” version that hasn’t been validated for your process. Before the delivery driver leaves and before you sign any shipping documents, take high-resolution photos of the nameplates of all critical components (motors, pumps, controllers) and the main equipment plate. If possible, power on the controller unit only to verify the firmware/software version on the boot-up screen. Cross-reference this against the version specified in your User Requirement Specification (URS) or purchase order. Rejecting the shipment at the door is far easier than dealing with the discrepancy later.
3. Installation and Connection Verification
This is the core of the IQ process, where the physical installation is scrutinized:

- Correct assembly and placement: verifying that the equipment is assembled and positioned as per the manufacturer’s instructions and engineering drawings.
- Utility connections: this is a critical step that involves confirming all connections to essential services. This includes checking that:
- Electrical: the power supply matches the required voltage and phase, and that proper grounding and safety circuits are in place.
- Plumbing: connections for water, steam, or drainage are correctly installed, leak-free, and made of the appropriate materials.
- Gases and compressed air: all pneumatic and gas lines are correctly connected, and the pressure and quality meet specifications.
- Ancillary equipment: ensuring any supporting or peripheral equipment is also correctly installed and connected.
- Software installation:for computer-controlled systems, this involves verifying that the software is installed correctly, communication with the...
You have read 14% of the article. The rest is for our community. Already a member? Log in
(and also to protect our original content from scraping bots)
Innovation.world community
Login or Register (100% free)
View the rest of this article and all members-only content and tools.
Only real engineers, manufacturers, designers, marketers professionals.
No bot, no hater, no spammer.
Related Readings
- Validation Master Plan (VMP): the high-level strategic document that defines the company’s overall validation philosophy, scope, responsibilities, and the specific systems and processes to be validated.
- User Requirement Specification (URS): the foundational document that details what the equipment or process is expected to do from an end-user and quality perspective, forming the basis for all subsequent testing.
- Factory Acceptance Testing (FAT) & Site Acceptance Testing (SAT): engineering-led precursor tests, often performed with the vendor, to verify equipment functionality before shipping (FAT) and after installation (SAT), which can be leveraged to streamline IQ/OQ.
- Computer System Validation (CSV): a specialized validation discipline that runs in parallel to IQ OQ PQ, focusing on the integrity, security, and reliability of the software and computerized systems controlling the equipment.
- 21 CFR Part 11 Compliance: the specific set of FDA regulations governing electronic records and electronic signatures, a critical component of CSV for ensuring data integrity, audit trails, and access control.
- Cleaning validation: a sometimes separated but related validation process performed on the same equipment to prove that a cleaning procedure can effectively and consistently remove product residues, cleaning agents, and microbial contamination (we strongly recommend to include it in the IQ OQ PQ protocol, as the cleaning status is an integral part of the product and process performances).
- Measurement System Analysis (MSA) / Gage R&R: the statistical methodology used to validate the *test methods* themselves, ensuring the measurement system used to judge product quality is accurate, precise, repeatable, and reproducible.
- Risk-based validation (using FMEA/HACCP): the methodology for identifying potential failure modes and risks to product quality, allowing validation efforts to be focused on the most Critical Process Parameters (CPPs).
- Critical Quality Attributes (CQAs) & Critical Process Parameters (CPPs): the specific, defined product characteristics (CQAs) and process variables (CPPs) that must be controlled to ensure the desired product quality, which form the acceptance criteria for PQ.
- Process Capability Analysis (Cpk/Ppk): the statistical analysis performed on PQ data to quantify how well a process is centered within its specification limits and how much variability it has, providing a numerical score of its “capability”.
- Continued Process Verification (CPV): the modern “Stage 3” of process validation, which involves the ongoing monitoring of process parameters and quality attributes during routine production to ensure the process remains in a constant state of control.
- Change control management: the formal quality system procedure for evaluating, documenting, and approving any proposed changes to a validated system or process to ensure it does not unintentionally exit its validated state.
- Deviation and CAPA management: the system for investigating, determining the root cause of, and implementing Corrective and Preventive Actions for any unexpected events or failures that occur during or after validation.
- Retrospective validation: an outdated validation approach, now rarely acceptable, based on analyzing historical production data from an existing, unvalidated process to attempt to prove it has been operating in a state of control.
- Validation Summary Report (VSR): the final, conclusive document that summarizes the entire validation effort (IQ OQ PQ), presents the key results, addresses any deviations, and formally declares the system or process as validated and fit for commercial use.
External Links on IQ OQ PQ Process Validation
International Standards
(hover the link to see our description of the content)
Glossary of Terms Used
American Society for Testing and Materials (ASTM): an international standards organization that develops and publishes voluntary consensus technical standards for materials, products, systems, and services, aimed at improving quality and safety across various industries.
Calculation of Process Capability (Cpk): a statistical measure that evaluates a process's ability to produce output within specified limits, calculated by assessing the distance between the process mean and the nearest specification limit, normalized by the process standard deviation.
Computer Numerically Controlled (CNC): a manufacturing process that uses programmed computer software to control machine tools, enabling precise and automated operation for tasks such as cutting, milling, drilling, and engraving materials.
Continuous Integration/Continuous Deployment (CI/CD): a software development practice that automates the integration of code changes and deployment to production environments, enabling frequent updates, faster delivery, and improved collaboration among development and operations teams through automated testing and monitoring.
Corrective Action and Preventative Action (CAPA): a systematic approach to identifying, investigating, and addressing nonconformities and potential issues to prevent recurrence and ensure compliance with regulatory standards in quality management systems.
Critical Control Points (CCP): specific stages in a process where control can be applied to prevent, eliminate, or reduce food safety hazards to acceptable levels. Identifying these points is essential for effective hazard analysis and critical control management in food production systems.
current Good Manufacturing Practice (cGMP): a system ensuring that products are consistently produced and controlled according to quality standards, encompassing regulations and guidelines for manufacturing processes, facilities, equipment, and personnel to ensure safety, quality, and efficacy in pharmaceuticals, food, and other regulated industries.
Defects Per Million Opportunities (DPMO): a measurement used in quality control that quantifies the number of defects in a process per one million opportunities for error, calculated by dividing the number of defects by the total number of opportunities and multiplying by one million.
Define Measure Analyze Improve Control (DMAIC): a data-driven quality strategy used in Six Sigma for process improvement, consisting of five phases: identifying the problem, measuring current performance, analyzing data to identify causes, improving processes based on findings, and controlling future performance to sustain improvements.
Design Validation (DV): a process to ensure a product meets specified requirements and intended use through testing and evaluation, confirming that the design fulfills its purpose and functions correctly in real-world conditions.
Failure Mode and Effects Analysis (FMEA): a systematic method for evaluating potential failure modes within a system, process, or product, assessing their effects on performance, and prioritizing risks to improve reliability and safety through corrective actions.
Food and Drug Administration (FDA): a federal agency of the United States Department of Health and Human Services responsible for regulating food safety, pharmaceuticals, medical devices, cosmetics, and tobacco products to ensure public health and safety through scientific evaluation and enforcement of compliance standards.
Good Manufacturing Practice (GMP): a system ensuring products are consistently produced and controlled according to quality standards, minimizing risks involved in pharmaceutical production and related industries. It encompasses guidelines for manufacturing processes, facility conditions, personnel qualifications, and documentation practices to ensure product safety and efficacy.
Hazard Analysis and Critical Control Points (HACCP): a systematic approach to food safety that identifies, evaluates, and controls hazards at critical points in the production process to prevent foodborne illnesses and ensure product safety.
Installation Qualification (IQ): a documented process to verify that equipment or systems are installed according to specifications, including assessment of utilities, environmental conditions, and compliance with design requirements, ensuring readiness for operational qualification.
Measurement System Analysis (MSA): a statistical method used to evaluate the accuracy, precision, and reliability of measurement processes and instruments, ensuring that data collected is valid and consistent for decision-making in quality control and process improvement.
Operational Qualification (OQ): a validation process that ensures equipment or systems operate according to specified requirements within defined limits, confirming that they perform as intended in their operational environment.
Performance Qualification (PQ): a process that verifies a system or equipment operates according to specified requirements under real-world conditions, ensuring it consistently performs its intended function within predetermined limits.
Process Capability Index (Cpk): a statistical measure that quantifies how well a process can produce output within specified limits, indicating the relationship between the process mean and the nearest specification limit, adjusted for process variability.
Process Performance Index (Ppk): a statistical measure that quantifies how well a process meets specification limits, calculated using the process mean and standard deviation. It indicates the capability of a process to produce output within defined limits, accounting for both variability and centering.
Product Lifecycle Management (PLM): a systematic approach to managing a product's lifecycle from inception, through engineering design and manufacturing, to service and disposal, integrating people, processes, data, and technology to improve product quality, reduce time to market, and enhance collaboration across stakeholders.
Production Part Approval Process (PPAP): a standardized procedure used in manufacturing to ensure that suppliers meet quality requirements before mass production, involving documentation and validation of design specifications, process capabilities, and production samples to confirm compliance with customer expectations.
Quality Management System (QMS): a structured system of processes, procedures, and responsibilities aimed at ensuring consistent quality in products and services, facilitating continuous improvement, and meeting customer and regulatory requirements.
Standard Operating Procedure (SOP): a set of step-by-step instructions created to help workers carry out routine operations consistently and efficiently, ensuring compliance with regulations and quality standards.
Statistical Process Control (SPC): a method of quality control that employs statistical techniques to monitor and control a process, ensuring it operates at its full potential by identifying variations and maintaining consistent output within specified limits.
User Requirement Specification (URS): a document detailing the needs and expectations of users for a system or product, outlining functional and non-functional requirements to guide development and ensure alignment with user objectives.
























Related Posts
How-to Best Fight a Pending Patent
All Patent Status: PCT vs Pending Patent vs Published Patent vs Granted Patent
Best 10 Patent Invalidation Strategies and Tools
Life Cycle Assessment (LCA) In Product Design Specifically
Product Value Analysis Overview
Workstation Ergonomic Assessment