Bien Fabricación Practice, or GMP, is the universal standard for quality production. It is a set of rules to ensure that products like medicines, food, and medical devices are made consistently and safely, batch after batch. The central idea is simple: quality cannot be inspected into a product at the end of the line. Instead, it must be built into every step of the manufacturing process, from the raw materials that arrive at the loading dock to the final package that leaves it.
The “c” in cGMP stands for Current. This single letter introduces a critical, dynamic requirement. While GMP provides the foundational rulebook, cGMP legally obligates manufacturers to use the most up-to-date tecnologías, systems, and scientific understanding available today. A process that was perfectly acceptable under GMP normas a decade ago might fail a cGMP inspection now if better, more reliable methods have since emerged. It forces companies to continuously improve.
Conclusiones Clave

- La distinción entre GMP y cGMP es ahora académica; la expectativa es universal.
- La gestión de riesgos de calidad (QRM) es el motor, no el papeleo ni el PLM.
- La integridad de los datos es un foco principal de auditoría.
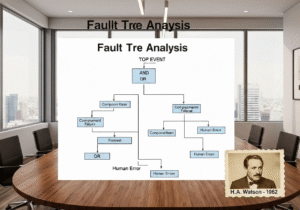
- El error humano es un síntoma, no la causa raíz. Atribuir una desviación a un error humano es una señal de alerta de un sistema de calidad deficiente.
- La Tecnología Analítica de Procesos (TAP) representa la transición de las pruebas a la garantía en tiempo real. La "c" de las buenas prácticas de fabricación actuales (cGMP) se ejemplifica con la TAP.
- La supervisión de los proveedores se basa en datos, no solo en auditorías.
- La Persona Cualificada (PC) representa una responsabilidad crítica específica de la UE.
- El Estrategia de control de la contaminación (CCS) es la nueva piedra angular de la fabricación estéril.
Los 10 principios básicos de las buenas prácticas de fabricación (BPF)
Las BPM no son solo un conjunto de normas, sino una mentalidad de calidad basada en diez principios fundamentales. Estos principios se combinan para crear un sistema sólido que garantiza la calidad integrada en cada etapa del producto, en lugar de simplemente evaluarse al final.

1. Redactar procedimientos e instrucciones de trabajo paso a paso
La base de las BPM es garantizar que todos los procesos estén claramente definidos y documentados. Este principio exige la creación de Procedimientos Operativos Estándar (POE) detallados e inequívocos para cada tarea crítica. El objetivo es garantizar que las operaciones se realicen de forma consistente y correcta en todo momento, independientemente de quién las realice. Esto elimina la ambigüedad y proporciona una referencia clara para la capacitación y la ejecución.
Ejemplo de aplicación: La empresa “PharmaBlend Inc.” fabrica un medicamento líquido sensible a la temperatura. Su POE para el “Control de Temperatura del Tanque de Preparación” (SOP-MFG-101) especifica no solo la temperatura objetivo (40 °C ± 2 °C), sino también la secuencia exacta para el encendido de la camisa de calentamiento, la velocidad de aumento de temperatura (sin superar los 5 °C por minuto), la sonda calibrada específica para la monitorización y las medidas a tomar si la temperatura sobrepasa los límites.
Consejo: En lugar de redactar POE monolíticos, utilice un enfoque modular. Cree POE "maestros" para procesos complejos que hagan referencia a documentos de instrucciones de trabajo más breves y específicos de cada tarea para cada paso (p. ej., calibrar un sensor específico, operar una válvula). Esto facilita las actualizaciones: si se reemplaza un solo equipo, solo necesita revisar una pequeña instrucción de trabajo en lugar de todo el POE del proceso, lo que reduce significativamente el tiempo de revisión y aprobación y minimiza el riesgo de introducir errores en secciones no relacionadas.
2. Siga los procedimientos e instrucciones meticulosamente

Tener procedimientos documentados no tiene sentido si no se siguen. Este principio exige un estricto cumplimiento de los POE escritos sin desviaciones. Si es necesario realizar una desviación, debe documentarse formalmente, justificarse y aprobarse mediante un proceso de control de cambios definido. Esto garantiza que cualquier desviación del estándar se controle, se evalúe su riesgo y se registre para su trazabilidad.
Consejo: Implemente una métrica "Correcto a la Primera" (RFT) para la ejecución de procedimientos, con seguimiento durante la revisión de registros de lotes. Cuando se produzcan desviaciones por incumplimiento, no se limite a capacitar nuevamente al operador. Realice un análisis de causa raíz centrado en la usabilidad del procedimiento (un enfoque de factores humanos). ¿La instrucción fue ambigua? ¿La secuencia fue ilógica? ¿Es difícil acceder a la herramienta requerida? Mejorar el procedimiento en sí mismo es una Acción Correctiva y Preventiva (CAPA) a largo plazo más eficaz que simplemente culpar al error humano.
3. Documentar el trabajo con prontitud y precisión

Este es el principio de que "si no se escribió, no ocurrió". Todas las actividades, desde la recepción de las materias primas hasta el envío del producto final, deben documentarse en tiempo real. Esto incluye el registro de datos, firmas, fechas y cualquier observación. Una documentación precisa y actualizada proporciona un historial completo y trazable de un lote (conocido como Registro de Lote o Registro del Historial del Dispositivo), esencial para investigar desviaciones, solucionar problemas y comprobar el cumplimiento durante una auditoría.
Consejo: Al diseñar registros de lotes (en papel o electrónicos), incorpore comprobaciones de integridad de datos directamente en los campos. Por ejemplo, en lugar de un espacio en blanco para la "Hora de finalización", estructúrelo para que requiera una hora de inicio y una hora de finalización, con una comprobación automatizada o manual para garantizar que la duración sea coherente con el paso del proceso. Para entradas críticas, utilice la verificación por segunda persona, pero asegúrese de que el verificador esté capacitado para ello. volver a realizar el cálculo crítico o verificar la configuración, no solo “marcar la casilla”.
Diferencia entre DMR y DHR:
- El registro maestro del dispositivo, o DMR, es la receta maestra para un dispositivo médicoEs una compilación formal y controlada de todas las instrucciones, especificaciones y procedimientos necesarios para producir un producto consistente. El DMR contiene todo, desde los planos de diseño y las especificaciones de los materiales hasta las instrucciones detalladas de fabricación, los métodos de control de calidad, el etiquetado y los requisitos de embalaje. Considérelo como el plano completo; define exactamente cómo debe fabricarse el dispositivo, de principio a fin.
- El registro del historial del dispositivo, o DHR, Es la prueba de que un lote, lote o unidad individual se fabricó según la receta. Es el registro de producción completo. El DHR contiene las fechas específicas de fabricación, las cantidades producidas, los resultados de las pruebas de ese lote e información de trazabilidad, como los números de serie o de lote. Si bien el DMR es el manual de instrucciones aplicable a todas las unidades, el DHR es la evidencia histórica que demuestra que una producción específica siguió esas instrucciones y cumplió con todos los criterios de aceptación.
4. Valide su trabajo y proceso

La validación es la prueba documentada de que un proceso, sistema o equipo produce sistemáticamente el resultado esperado. Este principio exige que los fabricantes demuestren que sus procesos son fiables...
Ha leído 14% del artículo. El resto es para nuestra comunidad. ¿Ya es miembro? Conectarse
(y también para proteger nuestro contenido original de los robots de scraping)
Comunidad.mundial.de.la.innovación
Iniciar sesión o registrarse (100% gratis)
Vea el resto de este artículo y todos los contenidos y herramientas exclusivos para miembros.
Sólo verdaderos ingenieros, fabricantes, diseñadores, profesionales del marketing.
Ni bot, ni hater, ni spammer.
Preguntas frecuentes
In a practical audit, how does an inspector’s expectation for ‘cGMP’ differ from the written ‘GMP’ normativa?
An inspector expects to see not just that you follow your written procedures (GMP), but that your procedures themselves reflect current industry best practices and technology (cGMP). They will question why you are using a 20-year-old analytical método when a more accurate and reliable one is now standard, or why you rely on manual checks where automated in-line verificación is now common. They are auditing your awareness and proactive adoption of modern quality standards.
¿CGMP significa que debemos invertir constantemente en la tecnología más nueva o podemos justificar el uso de equipos más antiguos y validados?
Puede justificar sin duda el uso de equipos antiguos, pero la carga de la prueba recae sobre usted. Su justificación debe estar documentada y basada en el riesgo. Debe demostrar mediante una validación sólida, un mantenimiento riguroso, una monitorización intensiva y datos de tendencias que su sistema antiguo proporciona un nivel de garantía de calidad equivalente o superior. control de procesos En comparación con las alternativas modernas, si su proceso con equipos antiguos presenta una mayor tasa de desviación, no podrá defenderlo.
Más allá de los registros de auditoría, ¿cuáles son las brechas de integridad de datos “ocultas” más comunes en las que se centran los reguladores?
Los organismos reguladores están examinando rigurosamente las hojas de cálculo no controladas que se utilizan para los cálculos de BPM, el uso de credenciales de inicio de sesión compartidas en equipos independientes (como balanzas o medidores de pH) y la posibilidad de realizar pruebas en equipos analíticos que pueden eliminarse sin dejar rastro. Otro aspecto importante es la integridad de los metadatos: la información sobre los datos, como las marcas de tiempo y los identificadores de usuario, que debe estar vinculada de forma segura al registro original.
¿En qué se diferencia un Sistema de Calidad Farmacéutica (PQS) según la ICH Q10 de simplemente tener un departamento de control de calidad sólido?
Un departamento de control de calidad fuerte hace cumplir calidad; un PQS gestiona como objetivo de toda la empresa. La diferencia clave es la integración formal de la alta dirección y el enfoque en rendimiento del proceso y la mejora continua. Un PQS garantiza que las métricas de calidad influyan directamente en las decisiones empresariales (como la asignación de recursos y la planificación estratégica) y que la dirección revise e impulse activamente la eficacia del sistema, en lugar de delegar todas las cuestiones de calidad en el control de calidad.
¿Qué significa realmente un “enfoque de ciclo de vida” para la validación de procesos (según ASTM E2500) para un ingeniero?
Significa que la validación ya no es un proceso de tres pasos. Es un proceso continuo. Para un ingeniero, esto significa:
- Etapa 1 (Diseño del proceso): utilizando Calidad por Diseño (QbD) para definir un proceso robusto y su espacio de control.
- Etapa 2 (Calificación del proceso): verificar que las instalaciones y los equipos sean adecuados para el propósito y que el proceso funcione consistentemente dentro de su espacio definido (PPQ).
- Etapa 3 (Verificación continua del proceso): Monitoreando activamente el proceso durante la producción rutinaria utilizando control estadístico de procesos (SPC) para garantizar que se mantenga en estado de control durante toda su vida comercial.
¿Cuál es la diferencia práctica más significativa entre las BPM de la UE (EudraLex) y las BPM de EE. UU.?
La diferencia más significativa es el papel de la persona cualificada (QP) en la UE. En EE.UU., la Unidad de Calidad tiene autoridad para autorizar un lote. En la UE, una QP nombrada específicamente debe certificar personalmente que cada lote ha sido fabricado y probado de acuerdo con todas las normativas y la marketing autorización antes de que pueda divulgarse. Esto hace recaer una inmensa responsabilidad personal y jurídica en un solo individuo.
Nuestro sistema CAPA cumple con las normas, pero los problemas suelen repetirse. ¿Cuál es la expectativa de las buenas prácticas de fabricación actuales (cGMP) para la eficacia de CAPA?
La expectativa de las cGMP es que verifique formalmente el funcionamiento de sus CAPA. Esto requiere incorporar una verificación de eficacia en su procedimiento CAPA. Esta verificación, realizada semanas o meses después de la implementación de la CAPA, debe proporcionar datos objetivos (p. ej., análisis de tendencias de las tasas de desviación, nuevos hallazgos de auditoría) para demostrar que se eliminó la causa raíz y que el problema no ha vuelto a ocurrir. Un CAPA cerrado sin esta verificación es una señal de alerta importante para los auditores.
¿Cómo ha evolucionado la expectativa de cGMP para la calificación de proveedores más allá de simplemente auditar al proveedor?
Las auditorías siguen siendo necesarias, pero las cGMP ahora exigen un enfoque más basado en datos y riesgos. Esto incluye el establecimiento de Acuerdos de Calidad formales que definan responsabilidades, la supervisión del desempeño del proveedor mediante métricas (p. ej., entrega puntual, tasas de desviación, calidad del material entrante) y la realización de análisis periódicos de materias primas para verificar el Certificado de Análisis (CoA) del proveedor. Debe demostrar una supervisión continua, no solo una certificación puntual.
Con la revisión del Anexo 1, ¿cuál es el cambio más grande en las cGMP en la fabricación estéril?
El cambio más importante es la exigencia de una Estrategia de Control de la Contaminación (ECC) formal e integral. Esta no se trata solo de un conjunto de POE, sino de un documento único e integral que justifica el diseño, los procesos y los programas de monitoreo de sus instalaciones con base en la gestión de riesgos. Le obliga a demostrar cómo todas sus medidas de control individuales (desde el uso de batas hasta la climatización y el diseño de procesos) funcionan en conjunto para prevenir la contaminación.
¿Por qué la Tecnología Analítica de Procesos (PAT) se considera un pilar de las cGMP modernas?
Porque PAT encarna el principio fundamental de las cGMP de incorporar la calidad, en lugar de probarla. Al proporcionar datos en tiempo real durante el proceso, PAT permite el control activo de los Parámetros Críticos del Proceso (PPC) para garantizar el cumplimiento de los Atributos Críticos de Calidad (ACC). Esto transforma la fabricación de un enfoque rígido y basado en recetas a un modelo flexible y basado en la ciencia, capaz de adaptarse a pequeñas variabilidades y garantizar un resultado consistente.
¿Cómo se debe tratar el “error humano” como causa raíz en un entorno de cGMP?
En un sistema cGMP maduro, el "error humano" rara vez es una causa raíz aceptable. Suele ser síntoma de un proceso o sistema defectuoso. Cuando ocurre un error, la investigación debe profundizar: ¿Fue confuso el procedimiento? ¿Fue inadecuada la capacitación? ¿El espacio de trabajo estaba mal diseñado (ingeniería de factores humanos)? ¿El operador estaba fatigado debido al exceso de horas extra? Un CAPA sólido abordará la falla subyacente del sistema, no solo volverá a capacitar al individuo.
Anual Reseña del producto (PQR) se considera a menudo una tarea. Cuál es su finalidad cGMP?
Su propósito es ser una herramienta proactiva para la mejora continua. El PQR no debe ser simplemente un volcado de datos retrospectivo. Es una oportunidad formal para analizar los datos de un año (tendencias, desviaciones, cambios, resultados de estabilidad) para evaluar el estado y la consistencia de un proceso. Su resultado más importante debe ser una lista de CAPA recomendadas y mejoras de proceso para el año siguiente.
Related Readings
- Calidad por diseño (QbD): un enfoque sistemático del desarrollo farmacéutico que hace hincapié en la garantía de calidad a lo largo de todo el proceso. ciclo de vida del producto.
- Diseño para Fabricabilidad (DFM): Técnicas para diseñar productos fáciles de fabricar, reduciendo costes y mejorando la calidad.
- Fabricación esbelta: principios orientados a minimizar el desperdicio y maximizar la productividad y la eficiencia en los procesos de fabricación.
- Six sigma: una metodología basada en datos enfocada en mejorar la calidad mediante la identificación y eliminación de causas de defectos en los procesos de fabricación.
- Gestión de riesgos: técnicas para identificar, evaluar y mitigar los riesgos en el diseño y la fabricación de productos, a menudo alineadas con ISO 14971 para productos sanitarios.
- Validación del proceso: métodos para confirmar que los procesos de fabricación producen consistentemente productos que cumplen con las especificaciones y los atributos de calidad predeterminados.
- Análisis de causa raíz (RCA): Técnicas para identificar las causas subyacentes de defectos o problemas en el proceso de fabricación.
- Failure Mode and Effects Analysis (FMEA): un enfoque estructurado para identificar posibles modos de falla en un producto o proceso y evaluar su impacto.
- Cumplimiento normativo: Comprender e implementar normas y regulaciones (por ejemplo, FDA, ISO) que rigen el diseño y la fabricación de productos.
- Gestión de la cadena de suministro: Estrategias para gestionar el flujo de materiales e información a través de la cadena de suministro para optimizar la eficiencia y la calidad.
- Control de cambios: procesos para gestionar cambios en productos o procesos en un entorno regulado para garantizar la coherencia y el cumplimiento.
- Control estadístico de procesos (CEP): Técnicas para monitorear y controlar un proceso a través de métodos estadísticos para mantener los niveles deseados de calidad.
- Sostenibilidad en la fabricación: métodos y prácticas destinados a reducir el impacto ambiental y mejorar la sostenibilidad de los procesos de fabricación.
Enlaces externos en
Normas internacionales
- ISO 22716:2007 Buenas Prácticas de Fabricación (BPF) - Directrices sobre Buenas Prácticas de Fabricación
- FDA 21 CFR Partes 210 y 211: Buenas prácticas de fabricación actuales en la fabricación, procesamiento, envasado o almacenamiento de medicamentos
- ISO 13485:2016 Productos sanitarios - Sistemas de gestión de calidad - Requisitos para fines reglamentarios
(Pase el cursor sobre el enlace para ver nuestra descripción del contenido)
Glosario de términos utilizados
American Society for Testing and Materials (ASTM): una organización internacional de normalización que desarrolla y publica normas técnicas de consenso voluntario para materiales, productos, sistemas y servicios, destinadas a mejorar la calidad y la seguridad en diversas industrias.
Application Programming Interface (API): un conjunto de reglas y protocolos que permite que diferentes aplicaciones de software se comuniquen e interactúen entre sí, posibilitando la integración de funcionalidades y el intercambio de datos entre sistemas.
Certificate of Analysis (CoA): un documento emitido por un fabricante o laboratorio de pruebas que confirma las especificaciones, la calidad y el cumplimiento de un producto con las normas reglamentarias, detallando los resultados de las pruebas y los métodos utilizados para el análisis.
Contamination Control Strategy (CCS): un enfoque sistemático para prevenir, detectar y mitigar la contaminación en entornos controlados, garantizando la calidad y seguridad del producto mediante procedimientos definidos, monitoreo y prácticas de gestión de riesgos.
Corrective Action and Preventative Action (CAPA): un enfoque sistemático para identificar, investigar y abordar no conformidades y problemas potenciales para prevenir su recurrencia y garantizar el cumplimiento de las normas regulatorias en los sistemas de gestión de calidad.
Critical Control Points (CCP): Etapas específicas de un proceso donde se puede aplicar control para prevenir, eliminar o reducir los riesgos para la inocuidad alimentaria a niveles aceptables. Identificar estos puntos es esencial para un análisis de riesgos eficaz y la gestión de controles críticos en los sistemas de producción de alimentos.
current Good Manufacturing Practice (cGMP): un sistema que garantiza que los productos se produzcan y controlen de manera consistente de acuerdo con estándares de calidad, abarcando regulaciones y pautas para procesos de fabricación, instalaciones, equipos y personal para garantizar la seguridad, la calidad y la eficacia en las industrias farmacéutica, alimentaria y otras industrias reguladas.
Device History Record (DHR): una compilación de registros que documenta el historial de producción de un dispositivo médico, incluidos los datos de fabricación, control de calidad y pruebas, garantizando el cumplimiento de los requisitos reglamentarios y facilitando la trazabilidad durante todo el ciclo de vida del dispositivo.
Device Master Record (DMR): una recopilación de documentos y especificaciones que proporcionan la información necesaria para producir un dispositivo médico, incluidas las especificaciones de diseño, los procesos de producción, las medidas de garantía de calidad y los requisitos de etiquetado, garantizando el cumplimiento de las normas regulatorias.
Failure Mode and Effects Analysis (FMEA): un método sistemático para evaluar modos de falla potenciales dentro de un sistema, proceso o producto, evaluar sus efectos sobre el desempeño y priorizar los riesgos para mejorar la confiabilidad y la seguridad a través de acciones correctivas.
Food and Drug Administration (FDA): una agencia federal del Departamento de Salud y Servicios Humanos de los Estados Unidos responsable de regular la seguridad alimentaria, los productos farmacéuticos, los dispositivos médicos, los cosméticos y los productos de tabaco para garantizar la salud y la seguridad públicas a través de la evaluación científica y la aplicación de las normas de cumplimiento.
Good Manufacturing Practice (GMP): Un sistema que garantiza la producción y el control constantes de los productos según los estándares de calidad, minimizando así los riesgos inherentes a la producción farmacéutica y las industrias afines. Abarca directrices para los procesos de fabricación, las condiciones de las instalaciones, la cualificación del personal y las prácticas de documentación para garantizar la seguridad y la eficacia de los productos.
Hazard Analysis and Critical Control Points (HACCP): un enfoque sistemático de la seguridad alimentaria que identifica, evalúa y controla los peligros en puntos críticos del proceso de producción para prevenir enfermedades transmitidas por los alimentos y garantizar la seguridad del producto.
Heating Ventilation and Air Conditioning (HVAC): Un sistema diseñado para regular el clima interior mediante el control de la temperatura, la humedad y la calidad del aire mediante procesos de calefacción, refrigeración y ventilación. Incluye componentes como hornos, aires acondicionados, conductos y termostatos para una gestión ambiental eficiente.
Installation Qualification (IQ): un proceso documentado para verificar que los equipos o sistemas estén instalados de acuerdo con las especificaciones, incluyendo la evaluación de los servicios públicos, las condiciones ambientales y el cumplimiento de los requisitos de diseño, garantizando la preparación para la calificación operativa.
International Organization for Standardization (ISO): Organismo internacional no gubernamental que desarrolla y publica normas para garantizar la calidad, la seguridad, la eficiencia y la interoperabilidad en diversas industrias y sectores, facilitando el comercio y la cooperación a nivel mundial. Fundado en 1947, está compuesto por organizaciones nacionales de normalización de los países miembros.
Key Performance Indicator (KPI): un valor medible que demuestra la eficacia con la que una organización está logrando objetivos comerciales clave, a menudo utilizado para evaluar el éxito en el logro de metas.
Magnetic Resonance Imaging (MRI): una técnica de imágenes médicas que utiliza fuertes campos magnéticos y ondas de radio para generar imágenes detalladas de las estructuras internas del cuerpo, particularmente de los tejidos blandos, mediante la detección de las señales emitidas por los núcleos de hidrógeno en presencia de un campo magnético.
Operational Qualification (OQ): un proceso de validación que garantiza que los equipos o sistemas funcionen de acuerdo con requisitos específicos dentro de límites definidos, confirmando que funcionan según lo previsto en su entorno operativo.
parts per million (ppm): Unidad de medida que representa la concentración de una sustancia en un millón de partes de otra, a menudo utilizada para cuantificar contaminantes en el aire, el agua o el suelo. Equivale a miligramos de sustancia por litro de solución o por kilogramo de material.
Performance Qualification (PQ): un proceso que verifica que un sistema o equipo funciona de acuerdo con requisitos específicos en condiciones del mundo real, garantizando que realiza consistentemente su función prevista dentro de límites predeterminados.
Product Lifecycle Management (PLM): un enfoque sistemático para gestionar el ciclo de vida de un producto desde su inicio, pasando por el diseño de ingeniería y la fabricación, hasta el servicio y la eliminación, integrando personas, procesos, datos y tecnología para mejorar la calidad del producto, reducir el tiempo de comercialización y mejorar la colaboración entre las partes interesadas.
Qualified Person (QP): una persona con la educación, la experiencia y la autoridad necesarias para supervisar y garantizar el cumplimiento de los requisitos reglamentarios en la preparación y presentación de documentos técnicos, en particular en los sectores de minería y recursos, según lo definen las normas industriales pertinentes.
Standard Operating Procedure (SOP): un conjunto de instrucciones paso a paso creadas para ayudar a los trabajadores a realizar operaciones rutinarias de manera consistente y eficiente, garantizando el cumplimiento de las regulaciones y los estándares de calidad.
Statistical Process Control (SPC): un método de control de calidad que emplea técnicas estadísticas para supervisar y controlar un proceso, garantizando que funcione a su máximo potencial identificando variaciones y manteniendo una producción constante dentro de límites específicos.