Risikobasierte Klassifizierung von Medizinprodukten
Medizinprodukte werden je nach Risiko für Patienten und Anwender in Klassen eingeteilt. Dieser risikobasierte Ansatz, der von Aufsichtsbehörden wie der US-amerikanischen FDA und in der EU verwendet wird, umfasst typischerweise drei oder vier Stufen (z. B. Klasse I, IIa, IIb, III). Geräte mit geringem Risiko wie Zungenspatel gehören zur Klasse I, während lebenserhaltende Geräte mit hohem Risiko wie Herzschrittmacher zur Klasse III gehören.
The risk-based classification system is a cornerstone of modern medical device regulation worldwide. It acknowledges that a one-size-fits-all regulatory approach is inefficient and potentially unsafe. Instead of subjecting every device to the most stringent review, regulators tailor their oversight to the potential for harm. The classification is determined by factors such as the device’s intended use, duration of contact with the body, degree of invasiveness, and whether it delivers energy or has a biological effect.
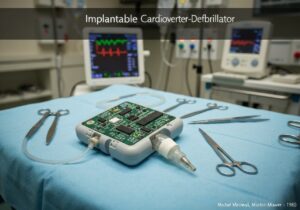
In den USA unterliegen beispielsweise Medizinprodukte der Klasse I (z. B. elastische Bandagen) den „Allgemeinen Kontrollen“, die die Registrierung und die korrekte Kennzeichnung umfassen. Medizinprodukte der Klasse II (z. B. Infusionspumpen, Elektrorollstühle) erfordern zusätzlich zu den allgemeinen Kontrollen „Spezielle Kontrollen“, die häufig Leistungsstandards und die Vorabbenachrichtigung (510(k)) beinhalten. Die höchste Risikokategorie, Klasse III (z. B. implantierbare Herzschrittmacher, Herzklappen), erfordert die strengste „Vorabzulassung“ (PMA), die die Vorlage umfangreicher klinischer Daten zum Nachweis von Sicherheit und Wirksamkeit beinhaltet.
Dieses gestaffelte System ermöglicht es den Aufsichtsbehörden, ihre Ressourcen auf Geräte mit dem größten Risiko zu konzentrieren und gleichzeitig den Marktzugang für risikoärmere Innovationen zu erleichtern. Das Konzept war grundlegend für die US-amerikanischen Medizinprodukteverordnungen von 1976 und wurde von Aufsichtsbehörden weltweit übernommen und angepasst. Es bildet die Basis für Rahmenbedingungen wie die Medizinprodukteverordnung (MDR) der Europäischen Union.
UNESCO Nomenclature: 3201
Öffentliche Gesundheit
Verwendung
Weitverbreitete Verwendung
Vorläufer
- Contergan-Tragödie (verdeutlichte die Notwendigkeit einer Arzneimittelregulierung, die sich auf die Geräteregulierung auswirkte)
- Dalkon-Shield-Vorfälle (ein Hauptgrund für die Änderungen des Medizinproduktegesetzes von 1976)
- Entwicklung allgemeiner Grundsätze des Risikomanagements im Ingenieurwesen
- Gründung nationaler Gesundheitsbehörden (z. B. FDA im Jahr 1906)
Anwendungen
- optimierte Zulassungsverfahren vor der Markteinführung für Geräte mit geringem Risiko
- abgestufte Anforderungen an die Überwachung nach dem Inverkehrbringen
- Internationale Bemühungen zur Harmonisierung der Regulierung (IMDRF)
- Entwicklung von Qualitätsmanagementsystemen wie ISO 13485, zugeschnitten auf das Risiko
Potenzielle Innovationsideen
Aufgrund des hohen Datenverkehrs durch Web-Scraping-Bots, der derzeit mehr als 40.000 Anfragen pro Tag umfasst, ist dieser Inhalt ausschließlich Community-Mitgliedern vorbehalten.
> Anmelden < oder > Registrieren < (100% kostenlos) Zugriff darauf sowie auf alle anderen eingeschränkten Inhalte und Tools.
Bezogen auf: Klassifizierung von Medizinprodukten, risikobasierte Regulierung, FDA-Klasse I, FDA-Klasse II, FDA-Klasse III, Zulassung vor dem Inverkehrbringen, 510(k), Medizinprodukteregulierung, öffentliche Gesundheit, Sicherheit.