Classificação de dispositivos médicos com base no risco
Os dispositivos médicos são classificados em classes com base no risco que representam para pacientes e usuários. Essa abordagem baseada no risco, utilizada por órgãos reguladores como o FDA (Food and Drug Administration) dos EUA e a UE (União Europeia), geralmente envolve três ou quatro níveis (por exemplo, Classe I, IIa, IIb, III). Dispositivos de baixo risco, como abaixadores de língua, são da Classe I, enquanto dispositivos de alto risco, essenciais para a vida, como marca-passos, são da Classe III.
The risk-based classification system is a cornerstone of modern medical device regulation worldwide. It acknowledges that a one-size-fits-all regulatory approach is inefficient and potentially unsafe. Instead of subjecting every device to the most stringent review, regulators tailor their oversight to the potential for harm. The classification is determined by factors such as the device’s intended use, duration of contact with the body, degree of invasiveness, and whether it delivers energy or has a biological effect.
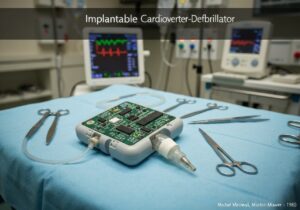
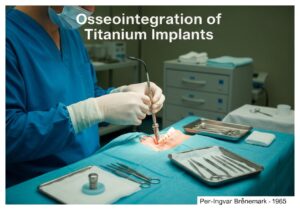
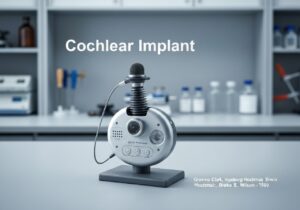
Por exemplo, nos Estados Unidos, dispositivos de Classe I (como bandagens elásticas) estão sujeitos a "Controles Gerais", que incluem registro e rotulagem adequada. Dispositivos de Classe II (como bombas de infusão e cadeiras de rodas motorizadas) exigem "Controles Especiais", além dos controles gerais, frequentemente incluindo padrões de desempenho e notificação pré-comercialização (510(k)). A categoria de maior risco, Classe III (como marca-passos implantáveis e válvulas cardíacas), exige a "Aprovação Pré-Comercialização" (PMA), a mais rigorosa, que envolve a apresentação de dados clínicos extensivos para comprovar a segurança e a eficácia.
Esse sistema hierárquico permite que as agências reguladoras concentrem seus recursos em dispositivos que representam o maior risco, ao mesmo tempo que facilita o acesso ao mercado para inovações de menor risco. O conceito foi fundamental para as Emendas de Dispositivos Médicos dos EUA de 1976 e foi adotado e adaptado por órgãos reguladores em todo o mundo, formando a base para estruturas como o Regulamento de Dispositivos Médicos (MDR) da União Europeia.
UNESCO Nomenclature: 3201
Saúde Pública
Precursores
- A tragédia da talidomida (evidenciou a necessidade de regulamentação de medicamentos, o que influenciou a regulamentação de dispositivos médicos)
- Incidentes com o dispositivo Dalkon Shield (um fator chave para as alterações de 1976 na legislação de dispositivos médicos)
- desenvolvimento de princípios gerais de gestão de riscos em engenharia
- estabelecimento de autoridades nacionais de saúde (ex.: FDA em 1906)
Aplicações
- Processos simplificados de aprovação pré-mercado para dispositivos de baixo risco
- requisitos de vigilância pós-comercialização em níveis
- esforços de harmonização regulatória internacional (IMDRF)
- development of quality management systems like iso 13485 tailored to risk
Ideias de Inovação Potencial
Devido ao tráfego de bots de coleta de dados, atualmente superior a 40 mil por dia, este conteúdo é reservado aos membros da comunidade.
> Login < ou > Registrar < (100% gratuito) para acessar isso, assim como todo o restante do conteúdo e das ferramentas restritas.
Relacionado a: classificação de dispositivos médicos, regulamentação baseada em risco, classe I da FDA, classe II da FDA, classe III da FDA, aprovação pré-comercialização, 510(k), regulamentação de dispositivos médicos, saúde pública, segurança.