
جيد تصنيع Practice, or GMP, is the universal standard for quality production. It is a set of rules to ensure that products like medicines, food, and medical devices are made consistently and safely, batch after batch. The central idea is simple: quality cannot be inspected into a product at the end of the line. Instead, it must be built into every step of the manufacturing process, from the raw materials that arrive at the loading dock to the final package that leaves it.
The “c” in cGMP stands for Current. This single letter introduces a critical, dynamic requirement. While GMP provides the foundational rulebook, cGMP legally obligates manufacturers to use the most up-to-date التقنيات, systems, and scientific understanding available today. A process that was perfectly acceptable under GMP المعايير a decade ago might fail a cGMP inspection now if better, more reliable methods have since emerged. It forces companies to continuously improve.
النقاط الرئيسية

- التمييز بين GMP و cGMP أصبح الآن أكاديميًا؛ والتوقع عالمي.
- إدارة مخاطر الجودة (QRM) هي المحرك، وليس الأوراق أو إدارة دورة حياة المنتج (PLM).
- تعتبر سلامة البيانات محورًا أساسيًا للتدقيق.
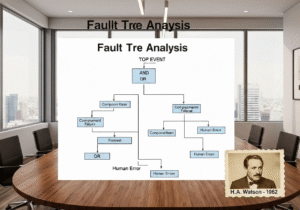
- "الخطأ البشري" مجرد عرض، وليس سببًا جذريًا. إن إسناد الانحراف إلى "خطأ بشري" يُعدّ مؤشرًا على ضعف نظام الجودة.
- تُجسّد تقنية تحليل العمليات (PAT) التحوّل من الاختبار إلى ضمان الجودة في الوقت الفعلي. ويُمثّل حرف "c" في cGMP مثالاً على ذلك في تقنية تحليل العمليات (PAT).
- إن الرقابة على الموردين تعتمد على البيانات، وليس فقط على التدقيق.
- يمثل الشخص المؤهل (QP) مسؤولية حاسمة خاصة بالاتحاد الأوروبي.
- ال استراتيجية مكافحة التلوث (CCS) is the new cornerstone of sterile manufacturing.
المبادئ الأساسية العشرة لممارسات التصنيع الجيدة (GMP)
ممارسات التصنيع الجيدة (GMP) ليست مجرد مجموعة قواعد، بل هي منهجية جودة مبنية على عشرة مبادئ أساسية. تتكامل هذه المبادئ لإنشاء نظام متين يضمن جودة المنتج في كل مرحلة، بدلاً من مجرد اختباره في النهاية.

1. اكتب الإجراءات وتعليمات العمل خطوة بخطوة
يرتكز أساس ممارسات التصنيع الجيدة (GMP) على ضمان وضوح وتوثيق جميع العمليات. يتطلب هذا المبدأ وضع إجراءات تشغيل قياسية (SOPs) مفصلة وواضحة لكل مهمة حيوية. الهدف هو ضمان تنفيذ العمليات بشكل متسق وصحيح في كل مرة، بغض النظر عن الجهة المنفذة. هذا يُزيل أي لبس ويوفر مرجعًا واضحًا للتدريب والتنفيذ.
مثال للتطبيق: تُصنّع شركة "فارما بلند" دواءً سائلاً حساسًا للحرارة. لا يقتصر إجراء التشغيل القياسي الخاص بها "بالتحكم في درجة حرارة خزان التركيب" (SOP-MFG-101) على تحديد درجة الحرارة المستهدفة (40 درجة مئوية ± درجتين مئويتين)، بل يشمل أيضًا التسلسل الدقيق لبدء تشغيل غلاف التسخين، ومعدل ارتفاع درجة الحرارة (لا يتجاوز 5 درجات مئوية في الدقيقة)، والمسبار المُعاير المُخصص للاستخدام للمراقبة، والإجراءات الواجب اتخاذها في حال تجاوز درجة الحرارة الحد الأقصى.
نصيحة: بدلاً من كتابة إجراءات تشغيلية قياسية متجانسة، استخدم نهجًا معياريًا. أنشئ إجراءات تشغيلية قياسية "رئيسية" للعمليات المعقدة، مع الإشارة إلى مستندات "تعليمات عمل" أصغر حجمًا ومحددة المهام لكل خطوة على حدة (مثل معايرة مستشعر معين، أو تشغيل صمام واحد). يتيح هذا تحديثات أسهل - ففي حال استبدال قطعة واحدة من المعدات، ستحتاج فقط إلى مراجعة تعليمات عمل صغيرة واحدة بدلاً من مراجعة إجراءات التشغيل القياسية للعملية بأكملها، مما يقلل بشكل كبير من وقت المراجعة والموافقة، ويقلل من خطر حدوث أخطاء في أقسام غير ذات صلة.
2. اتبع الإجراءات والتعليمات بدقة

لا جدوى من توثيق الإجراءات إذا لم تُتبع. يتطلب هذا المبدأ الالتزام الصارم بإجراءات التشغيل القياسية المكتوبة دون أي انحراف. إذا كان الانحراف ضروريًا، فيجب توثيقه رسميًا وتبريره والموافقة عليه من خلال عملية محددة لمراقبة التغيير. هذا يضمن مراقبة أي انحراف عن المعيار وتقييمه من حيث المخاطر وتسجيله لضمان إمكانية تتبعه.
نصيحة: تطبيق مقياس "الصواب من المرة الأولى" (RFT) لتنفيذ الإجراء، مع تتبعه أثناء مراجعة سجلات الدفعات. عند حدوث انحرافات نتيجة عدم الالتزام، لا تكتفِ بإعادة تدريب المُشغّل. بل أجرِ تحليلًا للسبب الجذري يُركّز على سهولة استخدام الإجراء (منهج العوامل البشرية). هل كانت التعليمات غامضة؟ هل كان التسلسل غير منطقي؟ هل يصعب الوصول إلى الأداة المطلوبة؟ يُعدّ تحسين الإجراء نفسه إجراءً تصحيحيًا ووقائيًا (CAPA) أكثر فعالية على المدى الطويل من مجرد إلقاء اللوم على الخطأ البشري.
3. توثيق العمل بسرعة ودقة

هذا هو مبدأ "ما لم يُدوّن، ما حدث". يجب توثيق جميع الأنشطة، من استلام المواد الخام إلى شحن المنتج النهائي، فورًا. يشمل ذلك تسجيل البيانات والتوقيعات والتواريخ وأي ملاحظات. يوفر التوثيق الدقيق والمتزامن تاريخًا كاملًا وقابلًا للتتبع للدفعة (يُعرف باسم سجل الدفعة أو سجل تاريخ الجهاز)، وهو أمر أساسي للتحقيق في الانحرافات، واستكشاف الأخطاء وإصلاحها، وإثبات الامتثال أثناء التدقيق.
نصيحة: عند تصميم سجلات الدفعات (ورقية أو إلكترونية)، أدرج "فحوصات سلامة البيانات" مباشرةً في الحقول. على سبيل المثال، بدلًا من مجرد مساحة فارغة لـ "وقت الانتهاء"، نظّمها بحيث تتطلب وقت بدء ووقت انتهاء، مع فحص آلي أو يدوي لضمان أن تكون المدة منطقية لخطوة العملية. بالنسبة للإدخالات المهمة، استخدم "التحقق بالثانية"، ولكن تأكد من تدريب المُتحقق على إعادة الأداء الحساب الحرج أو التحقق من الإعداد، وليس فقط "تحديد المربع".
الفرق بين DMR و DHR:
- سجل الجهاز الرئيسي، أو DMR، هي الوصفة الرئيسية لـ جهاز طبيهو تجميع رسمي ومُحكم لجميع التعليمات والمواصفات والإجراءات اللازمة لإنتاج منتج متناسق. يحتوي دليل المنتج النهائي (DMR) على كل شيء، بدءًا من رسومات التصميم ومواصفات المواد، وصولًا إلى تعليمات التصنيع المُفصّلة، وطرق اختبار مراقبة الجودة، ومتطلبات وضع العلامات والتغليف. يُمكن اعتباره بمثابة مخطط شامل؛ فهو يُحدد بدقة كيفية تصنيع الجهاز، من البداية إلى النهاية.
- سجل تاريخ الجهاز، أو DHR، هو الدليل على أن دفعة أو دفعة أو وحدة محددة قد صُنعت بالفعل وفقًا لتلك الوصفة. وهو سجل الإنتاج المكتمل. يحتوي سجل الإنتاج النهائي على تواريخ التصنيع المحددة، والكميات المُنتجة، ونتائج اختبارات تلك الدفعة، ومعلومات التتبع مثل الرقم التسلسلي أو رقم الدفعة. في حين أن سجل الإنتاج النهائي هو دليل التعليمات الذي ينطبق على جميع الوحدات، فإن سجل الإنتاج النهائي هو الدليل التاريخي الذي يُثبت أن دورة إنتاج واحدة قد اتبعت تلك التعليمات واستوفت جميع معايير القبول.
4. التحقق من صحة عملك وعمليتك

Validation is the documented proof that a process, system, or piece of equipment consistently produces the expected result. This principle requires manufacturers to prove that their processes are reliable...
You have read 14% of the article. The rest is for our community. Already a member? تسجيل الدخول
(وأيضًا لحماية المحتوى الأصلي لدينا من روبوتات الكشط)
مجتمع الابتكار العالمي
تسجيل الدخول أو التسجيل (100% مجاناً)
اطلع على بقية هذه المقالة وجميع المحتويات والأدوات الخاصة بالأعضاء فقط.
فقط المهندسون والمصنعون والمصممون والمسوقون الحقيقيون المحترفون.
لا روبوت، ولا كاره، ولا مرسل رسائل غير مرغوب فيها.
التعليمات
In a practical audit, how does an inspector’s expectation for ‘cGMP’ differ from the written ‘GMP’ اللوائح?
An inspector expects to see not just that you follow your written procedures (GMP), but that your procedures themselves reflect current industry best practices and technology (cGMP). They will question why you are using a 20-year-old analytical الطريقة when a more accurate and reliable one is now standard, or why you rely on manual checks where automated in-line تَحَقّق is now common. They are auditing your awareness and proactive adoption of modern quality standards.
هل يعني cGMP أنه يجب علينا الاستثمار باستمرار في أحدث التقنيات، أم يمكننا تبرير استخدام المعدات القديمة المعتمدة؟
يمكنك تبرير استخدام معدات قديمة بكل تأكيد، لكن عبء الإثبات يقع عليك. يجب أن يكون تبريرك موثقًا ومبنيًا على المخاطر. عليك إثبات أن نظامك القديم يوفر مستوىً مكافئًا أو أعلى من ضمان الجودة، وذلك من خلال عمليات تحقق دقيقة وصيانة دقيقة ومراقبة مكثفة وبيانات عن الاتجاهات. التحكم في العملية مقارنةً بالبدائل الحديثة. إذا كانت عمليتك التي تستخدم معدات قديمة ذات معدل انحراف أعلى، فلن تتمكن من حمايتها.
بعيدًا عن مسارات التدقيق، ما هي فجوات سلامة البيانات "غير المرئية" الأكثر شيوعًا والتي يركز عليها المنظمون؟
تُجري الجهات التنظيمية تدقيقًا مُكثّفًا لجداول البيانات غير المُراقَبة المُستخدمة في حسابات ممارسات التصنيع الجيدة (GMP)، واستخدام بيانات اعتماد تسجيل دخول مُشتركة على مُعدّات مُستقلة (مثل الموازين أو مقاييس الأس الهيدروجيني)، وإمكانية إجراء "عمليات تجريبية" على مُعدّات تحليلية يُمكن حذفها دون أي أثر. ومن النقاط الرئيسية الأخرى التي تُركّز عليها سلامة البيانات الوصفية - وهي البيانات المُتعلقة بالبيانات، مثل الطوابع الزمنية ومُعرّفات المُستخدمين، والتي يجب ربطها بشكل آمن بالسجل الأصلي.
كيف يختلف نظام الجودة الصيدلانية (PQS) بموجب ICH Q10 عن مجرد وجود قسم قوي لضمان الجودة؟
قسم ضمان الجودة القوي ينفذ الجودة؛ PQS يدير it as a business-wide objective. The key difference is the formal integration of senior management and a focus on أداء العملية and continuous improvement. A PQS ensures that quality metrics directly influence business decisions (like resource allocation and strategic planning) and that management is actively reviewing and driving the system’s effectiveness, as opposed to delegating all quality matters to QA.
ماذا يعني "نهج دورة الحياة" للتحقق من صحة العملية (وفقًا لمعيار ASTM E2500) بالنسبة للمهندس؟
هذا يعني أن التحقق لم يعد عمليةً مُتقطعةً، بل عمليةً مستمرة. بالنسبة للمهندس، هذا يعني:
- المرحلة 1 (تصميم العملية): استخدام الجودة من خلال التصميم (QbD) لتحديد عملية قوية ومساحة التحكم الخاصة بها.
- المرحلة الثانية (تأهيل العملية): التحقق من أن المنشأة والمعدات مناسبة للغرض وأن العملية تعمل باستمرار ضمن مساحتها المحددة (PPQ).
- المرحلة 3 (التحقق المستمر من العملية): مراقبة العملية بشكل نشط أثناء الإنتاج الروتيني باستخدام التحكم في العمليات الإحصائية (SPC) لضمان بقائها في حالة من السيطرة طوال حياتها التجارية.
ما هو الفرق العملي الأكثر أهمية بين EU GMP (EudraLex) و US cGMP؟
The most significant difference is the role of the Qualified Person (QP) in the EU. In the US, the Quality Unit has the authority to release a batch. In the EU, a specifically named QP must personally certify that each batch has been manufactured and tested in accordance with all regulations and the تسويق authorization before it can be released. This places an immense personal and legal responsibility on one individual.
نظامنا CAPA متوافق، ولكن المشاكل تتكرر باستمرار. ما هو توقع cGMP لفعالية CAPA؟
يتوقع cGMP أن تتحقق رسميًا من فعالية إجراءات CAPA. يتطلب هذا تضمين خطوة "فحص الفعالية" في إجراءات CAPA. يجب أن يوفر هذا الفحص، الذي يُجرى بعد أسابيع أو أشهر من تطبيق CAPA، بيانات موضوعية (مثل تحليل اتجاهات معدلات الانحراف، ونتائج التدقيق الجديدة) لإثبات القضاء على السبب الجذري وعدم تكرار المشكلة. يُعد إغلاق CAPA دون هذا التحقق علامة تحذير رئيسية للمدققين.
كيف تطورت توقعات cGMP لتأهيل الموردين إلى ما هو أبعد من مجرد التدقيق على الموردين؟
لا تزال عمليات التدقيق ضرورية، لكن ممارسات التصنيع الجيدة (cGMP) تتوقع الآن اتباع نهج أكثر اعتمادًا على البيانات ومراعاة المخاطر. يشمل ذلك إبرام اتفاقيات جودة رسمية تُحدد المسؤوليات، ومراقبة أداء المورد من خلال مقاييس (مثل: التسليم في الوقت المحدد، ومعدلات الانحراف، وجودة المواد الواردة)، وإجراء اختبارات دورية للمواد الخام للتحقق من شهادة تحليل المورد. يجب إثبات الإشراف المستمر، وليس مجرد تأهيل لمرة واحدة.
مع مراجعة الملحق 1، ما هو التحول الأكبر في ممارسات التصنيع الجيدة الحالية (cGMP) في مجال التصنيع المعقم؟
يتمثل التحول الأكبر في ضرورة وضع استراتيجية رسمية وشاملة لمكافحة التلوث (CCS). هذه الاستراتيجية ليست مجرد مجموعة من الإجراءات التشغيلية القياسية، بل هي وثيقة شاملة تُبرر تصميم منشأتك وعملياتها وبرامج المراقبة القائمة على إدارة المخاطر. تُلزمك هذه الاستراتيجية بتوضيح كيفية عمل جميع تدابير التحكم الفردية (من ارتداء الملابس الواقية إلى أنظمة التدفئة والتهوية وتكييف الهواء وتصميم العمليات) معًا لمنع التلوث.
لماذا تعتبر تقنية التحليل العملي (PAT) أحد ركائز ممارسات التصنيع الجيدة الحديثة (cGMP)؟
لأن PAT يُجسّد مبدأ cGMP الأساسي المتمثل في بناء الجودة، بدلاً من اختبارها. من خلال توفير بيانات آنية أثناء العملية، يُتيح PAT التحكم النشط في معايير العملية الحرجة (CPPs) لضمان استيفاء سمات الجودة الحرجة (CQA). هذا يُحوّل التصنيع من نهج صارم قائم على الوصفات إلى نموذج مرن قائم على العلم، قادر على التكيف مع المتغيرات الطفيفة وضمان نتيجة ثابتة.
كيف ينبغي التعامل مع "الخطأ البشري" باعتباره السبب الجذري في بيئة cGMP؟
في نظام cGMP ناضج، نادرًا ما يكون "الخطأ البشري" سببًا جذريًا مقبولًا. فهو عادةً ما يكون مؤشرًا على خلل في العملية أو النظام. عند حدوث خطأ، يجب التعمق في التحقيق: هل كان الإجراء مُربكًا؟ هل كان التدريب غير كافٍ؟ هل كان تصميم مكان العمل سيئًا (هندسة العوامل البشرية)؟ هل أُرهق المُشغّل بسبب العمل الإضافي المفرط؟ سيُعالج برنامج CAPA القوي عطل النظام الأساسي، وليس مجرد إعادة تدريب الفرد.
The Annual مراجعة المنتج (PQR) is often seen as a chore. What is its intended cGMP purpose?
الغرض من هذا التقرير هو أن يكون أداة استباقية للتحسين المستمر. لا ينبغي أن يكون تقرير جودة العمليات (PQR) مجرد تفريغ بيانات بأثر رجعي، بل هو فرصة رسمية لتحليل بيانات عام كامل (الاتجاهات، الانحرافات، التغييرات، نتائج الاستقرار) لتقييم سلامة واتساق العملية. وينبغي أن تكون أهم مخرجاته قائمةً بإجراءات التقييم والتحسينات التصحيحية (CAPAs) الموصى بها وتحسينات العمليات للعام المقبل.
قراءات ذات صلة
- الجودة من خلال التصميم (QbD): a systematic approach to pharmaceutical development that emphasizes quality assurance throughout the دورة حياة المنتج.
- التصميم من أجل القدرة على التصنيع (DFM): تقنيات لتصميم المنتجات التي يسهل تصنيعها، مما يقلل التكاليف ويعزز الجودة.
- التصنيع المرن: المبادئ التي تهدف إلى تقليل النفايات مع تعظيم الإنتاجية والكفاءة في عمليات التصنيع.
- Six sigma: منهجية تعتمد على البيانات تركز على تحسين الجودة من خلال تحديد وإزالة أسباب العيوب في عمليات التصنيع.
- إدارة المخاطر: techniques for identifying, assessing, and mitigating risks in product design and manufacturing, often aligned with ايزو 14971 for medical devices.
- التحقق من صحة العملية: طرق للتأكد من أن عمليات التصنيع تنتج باستمرار منتجات تلبي المواصفات وسمات الجودة المحددة مسبقًا.
- تحليل السبب الجذري (RCA): تقنيات لتحديد الأسباب الكامنة وراء العيوب أو المشاكل في عملية التصنيع.
- Failure Mode and Effects Analysis (FMEA): نهج منظم لتحديد أنماط الفشل المحتملة في المنتج أو العملية وتقييم تأثيرها.
- الامتثال التنظيمي: فهم وتنفيذ المعايير واللوائح (على سبيل المثال، FDA و ISO) التي تحكم تصميم المنتج وتصنيعه.
- إدارة سلسلة التوريد: استراتيجيات لإدارة تدفق المواد والمعلومات عبر سلسلة التوريد لتحسين الكفاءة والجودة.
- التحكم في التغيير: عمليات إدارة التغييرات في المنتجات أو العمليات في بيئة منظمة لضمان الاتساق والامتثال.
- التحكم الإحصائي في العملية (SPC): تقنيات لمراقبة والتحكم في عملية ما من خلال الأساليب الإحصائية للحفاظ على مستويات الجودة المطلوبة.
- الاستدامة في التصنيع: الأساليب والممارسات التي تهدف إلى تقليل الأثر البيئي وتعزيز استدامة عمليات التصنيع.
الروابط الخارجية على
المعايير الدولية
(حرك الرابط لرؤية وصفنا للمحتوى)
مسرد المصطلحات المستخدمة
American Society for Testing and Materials (ASTM): منظمة معايير دولية تعمل على تطوير ونشر المعايير الفنية الطوعية المتفق عليها للمواد والمنتجات والأنظمة والخدمات، بهدف تحسين الجودة والسلامة في مختلف الصناعات.
Application Programming Interface (API): مجموعة من القواعد والبروتوكولات التي تسمح لتطبيقات البرامج المختلفة بالتواصل والتفاعل مع بعضها البعض، مما يتيح تكامل الوظائف وتبادل البيانات بين الأنظمة.
Certificate of Analysis (CoA): وثيقة صادرة عن الشركة المصنعة أو مختبر الاختبار تؤكد مواصفات المنتج وجودته وامتثاله للمعايير التنظيمية، وتفصل نتائج الاختبار والطرق المستخدمة للتحليل.
Contamination Control Strategy (CCS): نهج منهجي لمنع التلوث واكتشافه وتخفيفه في البيئات الخاضعة للرقابة، وضمان جودة المنتج وسلامته من خلال إجراءات محددة وممارسات المراقبة وإدارة المخاطر.
Corrective Action and Preventative Action (CAPA): نهج منهجي لتحديد حالات عدم المطابقة والمشكلات المحتملة والتحقيق فيها ومعالجتها لمنع تكرارها وضمان الامتثال للمعايير التنظيمية في أنظمة إدارة الجودة.
Critical Control Points (CCP): مراحل محددة في العملية، حيث يُمكن تطبيق الرقابة لمنع أو القضاء على أو تقليل مخاطر سلامة الغذاء إلى مستويات مقبولة. يُعدّ تحديد هذه النقاط أساسيًا لتحليل المخاطر بفعالية وإدارة الرقابة الحرجة في أنظمة إنتاج الغذاء.
current Good Manufacturing Practice (cGMP): نظام يضمن إنتاج المنتجات والتحكم فيها باستمرار وفقًا لمعايير الجودة، ويشمل اللوائح والمبادئ التوجيهية لعمليات التصنيع والمرافق والمعدات والموظفين لضمان السلامة والجودة والفعالية في الصناعات الدوائية والغذائية وغيرها من الصناعات المنظمة.
Device History Record (DHR): مجموعة من السجلات التي توثق تاريخ إنتاج جهاز طبي، بما في ذلك بيانات التصنيع ومراقبة الجودة والاختبار، مما يضمن الامتثال للمتطلبات التنظيمية وتسهيل إمكانية التتبع طوال دورة حياة الجهاز.
Device Master Record (DMR): مجموعة من الوثائق والمواصفات التي توفر المعلومات اللازمة لإنتاج جهاز طبي، بما في ذلك مواصفات التصميم وعمليات الإنتاج وتدابير ضمان الجودة ومتطلبات وضع العلامات، وضمان الامتثال للمعايير التنظيمية.
Failure Mode and Effects Analysis (FMEA): طريقة منهجية لتقييم أوضاع الفشل المحتملة داخل النظام أو العملية أو المنتج، وتقييم آثارها على الأداء، وإعطاء الأولوية للمخاطر لتحسين الموثوقية والسلامة من خلال الإجراءات التصحيحية.
Food and Drug Administration (FDA): وكالة اتحادية تابعة لوزارة الصحة والخدمات الإنسانية الأمريكية مسؤولة عن تنظيم سلامة الأغذية والأدوية والأجهزة الطبية ومستحضرات التجميل ومنتجات التبغ لضمان الصحة العامة والسلامة من خلال التقييم العلمي وإنفاذ معايير الامتثال.
Good Manufacturing Practice (GMP): نظام يضمن إنتاج المنتجات ومراقبتها باستمرار وفقًا لمعايير الجودة، مما يقلل من المخاطر المرتبطة بإنتاج الأدوية والصناعات ذات الصلة. ويشمل إرشادات لعمليات التصنيع، وظروف المنشأة، ومؤهلات الموظفين، وممارسات التوثيق لضمان سلامة المنتج وفعاليته.
Hazard Analysis and Critical Control Points (HACCP): نهج منهجي لسلامة الغذاء يعمل على تحديد المخاطر وتقييمها والسيطرة عليها عند نقاط حرجة في عملية الإنتاج لمنع الأمراض المنقولة بالغذاء وضمان سلامة المنتج.
Heating Ventilation and Air Conditioning (HVAC): نظام مصمم لتنظيم مناخ المنزل من خلال التحكم في درجة الحرارة والرطوبة وجودة الهواء من خلال عمليات التدفئة والتبريد والتهوية. ويشمل مكونات مثل الأفران ومكيفات الهواء وقنوات التهوية ومنظمات الحرارة لضمان إدارة بيئية فعّالة.
Installation Qualification (IQ): عملية موثقة للتحقق من تركيب المعدات أو الأنظمة وفقًا للمواصفات، بما في ذلك تقييم المرافق والظروف البيئية والامتثال لمتطلبات التصميم، وضمان الجاهزية للتأهيل التشغيلي.
International Organization for Standardization (ISO): هيئة دولية غير حكومية تُعنى بتطوير ونشر المعايير لضمان الجودة والسلامة والكفاءة والتوافق التشغيلي في مختلف الصناعات والقطاعات، مما يُسهّل التجارة والتعاون العالميين. تأسست عام ١٩٤٧، وتضم منظمات التقييس الوطنية من الدول الأعضاء.
Key Performance Indicator (KPI): قيمة قابلة للقياس توضح مدى فعالية المنظمة في تحقيق الأهداف التجارية الرئيسية، وغالبًا ما تستخدم لتقييم النجاح في الوصول إلى الأهداف.
Magnetic Resonance Imaging (MRI): تقنية تصوير طبي تستخدم مجالات مغناطيسية قوية وموجات راديوية لتوليد صور مفصلة للهياكل الداخلية للجسم، وخاصة الأنسجة الرخوة، من خلال اكتشاف الإشارات المنبعثة من نوى الهيدروجين في وجود مجال مغناطيسي.
Operational Qualification (OQ): عملية التحقق التي تضمن أن المعدات أو الأنظمة تعمل وفقًا للمتطلبات المحددة ضمن حدود محددة، مما يؤكد أنها تعمل كما هو مقصود في بيئة التشغيل الخاصة بها.
parts per million (ppm): وحدة قياس تُمثل تركيز مادة ما في مليون جزء من مادة أخرى، وتُستخدم غالبًا لتحديد كمية الملوثات في الهواء أو الماء أو التربة. وهي تعادل مليغرامات المادة لكل لتر من المحلول أو لكل كيلوغرام من المادة.
Performance Qualification (PQ): عملية تتحقق من أن النظام أو المعدات تعمل وفقًا لمتطلبات محددة في ظل ظروف العالم الحقيقي، مما يضمن قيامها بوظيفتها المقصودة باستمرار ضمن حدود محددة مسبقًا.
Product Lifecycle Management (PLM): نهج منهجي لإدارة دورة حياة المنتج من البداية، من خلال التصميم الهندسي والتصنيع، إلى الخدمة والتخلص منها، ودمج الأشخاص والعمليات والبيانات والتكنولوجيا لتحسين جودة المنتج وتقليل الوقت المستغرق لطرح المنتج في السوق وتعزيز التعاون بين أصحاب المصلحة.
Qualified Person (QP): فرد يتمتع بالتعليم والخبرة والسلطة اللازمة للإشراف على الامتثال للمتطلبات التنظيمية وضمانه في إعداد وتقديم الوثائق الفنية، وخاصة في قطاعي التعدين والموارد، كما هو محدد بواسطة معايير الصناعة ذات الصلة.
Standard Operating Procedure (SOP): مجموعة من التعليمات خطوة بخطوة تم إنشاؤها لمساعدة العمال على تنفيذ العمليات الروتينية باستمرار وبكفاءة، وضمان الامتثال للوائح ومعايير الجودة.
Statistical Process Control (SPC): طريقة لمراقبة الجودة تستخدم تقنيات إحصائية لمراقبة عملية ما والتحكم فيها، والتأكد من أنها تعمل بكامل إمكاناتها من خلال تحديد الاختلافات والحفاظ على الناتج المتسق ضمن حدود محددة.
























منشورات ذات صلة
النص الكامل لتسميات اليونسكو
استراتيجية مكافحة التلوث والغرف النظيفة: 26 أفضل الممارسات
التحقق من صحة عملية IQ OQ PQ: النظرية الكاملة والتطبيق العملي
استراتيجيات "الجوز الوحيد"، و"التابع الأول"، و"التابع السريع"
أفضل 20 استخدامًا للوكلاء في الهندسة
كيفية بيع الثلج للإسكيمو (أو بالأحرى حيل التسويق)