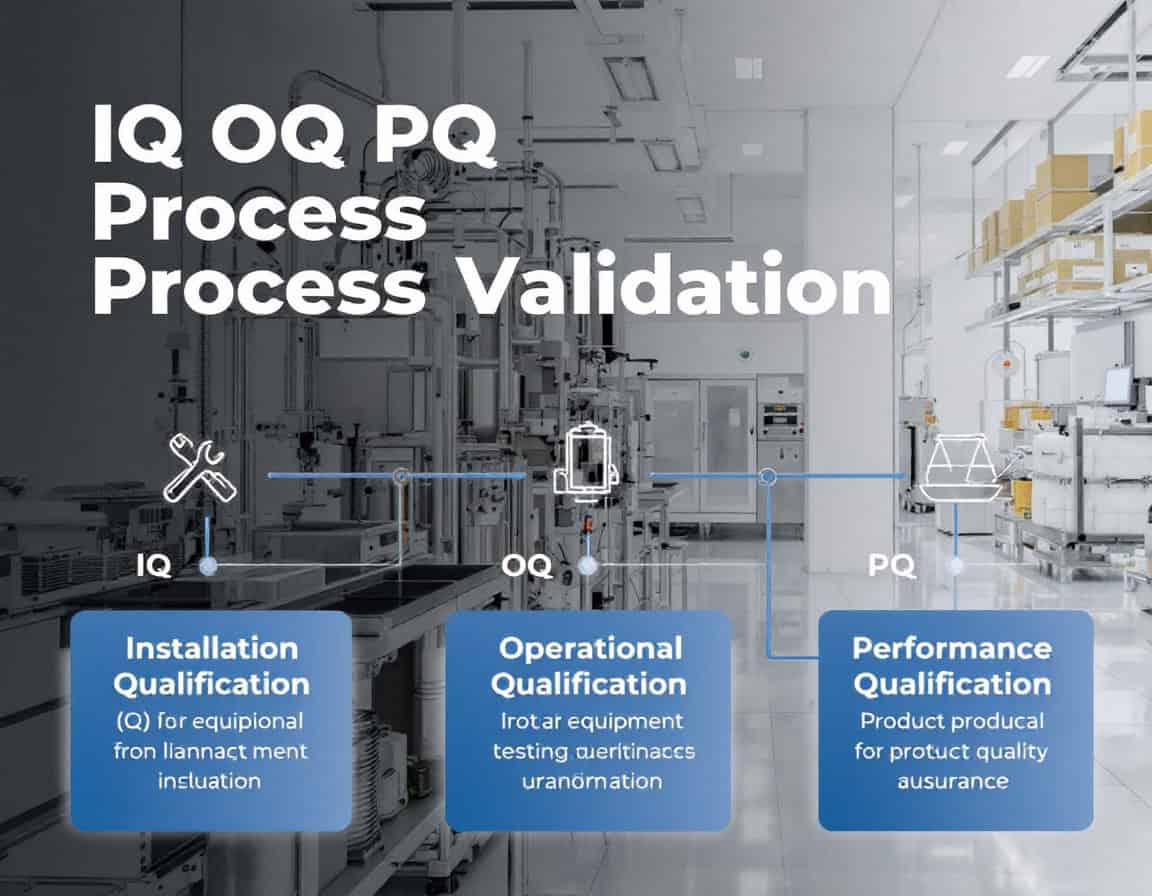
La metodología de validación del proceso IQ OQ PQ, que significa Calificación de Instalación (IQ), Calificación Operacional (OQ) y Calificación de Desempeño (PQ), constituye la secuencia establecida estructura Para la validación de procesos, se utiliza la generación sistemática de evidencia documentada de que un proceso de fabricación se encuentra bajo control. Esta metodología sigue una progresión deliberada y lógica, comenzando con la verificación estática y documentada de la correcta instalación del equipo (IQ), pasando a la confirmación dinámica de su funcionamiento fiable en todos sus rangos especificados (OQ), y culminando con la demostración de que el proceso integrado produce productos que cumplen sistemáticamente con todos los atributos de calidad (PQ).
Toda la secuencia está diseñada para proporcionar una prueba objetiva y rastreable de que el proceso es sólido, reproducible y adecuado para el propósito previsto antes de su uso comercial.
Esta metodología es un requisito regulatorio no negociable en industrias regidas por cGMP, como productos farmacéuticos, biológicos y dispositivo médico Fabricación, donde la consistencia del proceso está directamente relacionada con la seguridad del paciente. Su aplicación no se limita a la investigación inicial, sino a la etapa crítica de validación que se lleva a cabo una vez finalizado el desarrollo del proceso y antes de su aprobación para la producción comercial rutinaria.
Conclusiones Clave

- El resultado de la validación es evidencia, no producto.
- Las pruebas OQ del “peor caso” implican una combinación de factores estresantes.
- PQ valida el proceso integrado, no sólo el equipo.
- La regla de los “tres lotes” demuestra reproducibilidad.
- El éxito de PQ se define por la constancia, no solo por aprobar las especificaciones.
- El protocolo es un contrato; las desviaciones deben estar justificadas y bien documentadas.
- Aproveche la documentación del proveedor (FAT/SAT), pero no reemplace su propia OQ
- La validación define el inicio del ciclo de vida, no el final de un proyecto.
- Cualquier cambio “igual por igual” requiere justificación.
- La capacitación del personal es un requisito previo para OQ y PQ
Calificación de instalación (IQ)
La Calificación de la Instalación (IQ) es la fase inicial y fundamental de la metodología de validación del proceso IQ OQ PQ. Es un proceso formal y documentado que verifica y confirma que los equipos, sistemas y componentes auxiliares se han entregado, instalado y configurado en total conformidad con las recomendaciones del fabricante y las especificaciones de diseño del usuario.
El principio fundamental de IQ es proporcionar evidencia documentada de que la instalación es correcta y crea un entorno adecuado y seguro para las fases de calificación posteriores.
El propósito y la importancia del CI: El objetivo principal de IQ es establecer una línea base para la validación del equipo. Antes de comenzar cualquier prueba operativa, es crucial garantizar que el equipo esté físicamente presente, correctamente ensamblado y ubicado en un entorno adecuado. Esta fase mitiga los riesgos asociados a una instalación incorrecta, que de otro modo podrían provocar fallos en el equipo, problemas de calidad del producto y riesgos de seguridad.
Etapas y actividades clave en la calificación de la instalación
El proceso IQ es sistemático e implica varias etapas detalladas, normalmente gestionadas a través de un protocolo y una lista de verificación previamente aprobados.
1. Preinstalación y planificación

Antes incluso de que el equipo llegue o se solicite, el proceso de IQ comienza con una planificación minuciosa. Esto incluye:
- Verificación de preparación del sitio: Asegurarse de que la ubicación designada esté preparada para el nuevo equipo. Esto implica verificar la superficie adecuada, el soporte estructural y la disponibilidad de los servicios públicos necesarios en los puntos de conexión correctos.
- Condiciones ambientales: verificar que el entorno de instalación cumpla con las especificaciones del fabricante en cuanto a temperatura, humedad y limpieza.
- Recopilación de documentación: recopilar todos los documentos esenciales, como la orden de compra, los manuales del fabricante, las especificaciones de diseño, los dibujos de ingeniería y los certificados de calibración de cualquier instrumento de medición integrado.
Consejo: Recorra el recorrido y modele el volumen. No se limite a mirar un plano 2D. Los fallos de IQ más comunes y costosos ocurren aquí. Recorra literalmente la ruta planificada desde el muelle de carga hasta el punto de instalación final con las partes interesadas clave (Instalaciones, Ingeniería y el jefe de proyecto del proveedor). Utilice un marco sencillo de madera o PVC construido con las dimensiones máximas de altura, anchura y longitud de la máquina. Lleve físicamente este marco "fantasma" a lo largo de todo el recorrido. Esto revelará problemas de espacio libre con puertas, tuberías bajas, esquinas estrechas y la capacidad de los ascensores que los planos suelen pasar por alto. Además, verifique la capacidad de carga del piso no sólo en el punto final, sino a lo largo de todo el recorrido de tránsito.

2. Recepción y verificación del equipo
Una vez entregado el equipo se realiza una inspección exhaustiva:
- Verificación de componentes: Los artículos entregados se verifican meticulosamente comparándolos con la lista de embalaje y la orden de compra para confirmar que se han recibido todos los componentes, incluido el software, y que son el modelo y la versión correctos solicitados.
- Inspección de daños: Se realiza una inspección visual para garantizar que no se hayan producido daños durante el envío y la manipulación.
Nota: En la mayoría de las industrias que aplican la metodología IQ OQ PQ, el equipo debe incluir sus certificados de conformidad (marcado CE, FDA, etc.), eventualmente otros certificados y, posiblemente, los resultados de pruebas propias del fabricante. Estos documentos deben considerarse con la misma importancia que los propios productos físicos. De hecho, se debe rechazar una entrega si estos documentos no se incluyen o no se reciben con antelación (al menos en la zona de cuarentena).
Consejo relacionado: Confíe, pero verifique el firmware. La lista de empaque es lo mínimo indispensable. Debe centrarse en los componentes críticos y, sobre todo, en las versiones de software y firmware. Un proveedor podría enviar una versión más reciente y "mejor" que no haya sido validada para su proceso. Antes de que el conductor se marche y de firmar cualquier documento de envío, tome fotos de alta resolución de las placas de identificación de todos los componentes críticos (motores, bombas, controladores) y de la placa del equipo principal. Si es posible, encienda la unidad del controlador solo para verificar la versión de firmware/software en la pantalla de arranque. Compare esta información con la versión especificada en su Especificación de Requisitos del Usuario (URS) o en la orden de compra. Rechazar el envío en la puerta es mucho más fácil que solucionar la discrepancia posteriormente.
3. Instalación y verificación de la conexión
Este es el núcleo del proceso IQ, donde se examina la instalación física:

- Montaje y colocación correctos: verificar que el equipo esté ensamblado y posicionado según las instrucciones del fabricante y los dibujos de ingeniería.
- Conexiones de servicios públicos: Este es un paso crucial que implica confirmar todas las conexiones a los servicios esenciales. Esto incluye verificar que:
- Eléctrico: la fuente de alimentación coincide con el voltaje y la fase requeridos, y que existen circuitos de seguridad y conexión a tierra adecuados.
- Plomería: Las conexiones de agua, vapor o drenaje están correctamente instaladas, no tienen fugas y están hechas de los materiales adecuados.
- Gases y aire comprimido: Todas las líneas neumáticas y de gas están conectadas correctamente y la presión y la calidad cumplen con las especificaciones.
- Equipos auxiliares: asegurándose de que cualquier equipo de soporte o periférico también esté correctamente instalado y conectado.
- Instalación del software:for computer-controlled systems, this involves verifying that the software is installed correctly, communication with the...
You have read 14% of the article. The rest is for our community. Already a member? Conectarse
(y también para proteger nuestro contenido original de los robots de scraping)
Comunidad.mundial.de.la.innovación
Iniciar sesión o registrarse (100% gratis)
Vea el resto de este artículo y todos los contenidos y herramientas exclusivos para miembros.
Sólo verdaderos ingenieros, fabricantes, diseñadores, profesionales del marketing.
Ni bot, ni hater, ni spammer.
Related Readings
- Plan Maestro de Validación (PMV): el documento estratégico de alto nivel que define la filosofía general de validación de la empresa, el alcance, las responsabilidades y los sistemas y procesos específicos que se validarán.
- Especificación de requisitos del usuario (URS): el documento fundamental que detalla lo que se espera que haga el equipo o proceso desde una perspectiva de calidad y del usuario final, y que constituye la base para todas las pruebas posteriores.
- Pruebas de aceptación en fábrica (FAT) y pruebas de aceptación en sitio (SAT): pruebas precursoras dirigidas por ingeniería, a menudo realizadas con el proveedor, para verificar la funcionalidad del equipo antes del envío (FAT) y después de la instalación (SAT), que se pueden aprovechar para optimizar la IQ/OQ.
- Validación del sistema informático (CSV): una disciplina de validación especializada que se ejecuta en paralelo a IQ OQ PQ y se centra en la integridad, seguridad y confiabilidad del software y los sistemas computarizados que controlan el equipo.
- Cumplimiento de la Parte 11 del Título 21 del CFR: el conjunto específico de regulaciones de la FDA que rigen los registros electrónicos y las firmas electrónicas, un componente crítico de CSV para garantizar la integridad de los datos, los registros de auditoría y el control de acceso.
- Validación de limpieza: un proceso de validación a veces separado pero relacionado, realizado en el mismo equipo para demostrar que un procedimiento de limpieza puede eliminar de manera efectiva y consistente los residuos del producto, los agentes de limpieza y la contaminación microbiana (recomendamos enfáticamente incluirlo en el protocolo IQ OQ PQ, ya que el estado de limpieza es una parte integral del rendimiento del producto y del proceso).
- Análisis del sistema de medición (MSA) / Gage R&R: la metodología estadística utilizada para validar los *métodos de prueba* en sí, garantizando que el sistema de medición utilizado para juzgar la calidad del producto sea preciso, exacto, repetible y reproducible.
- Validación basada en riesgos (utilizando FMEA/HACCP): la metodología para identificar modos de falla potenciales y riesgos para la calidad del producto, permitiendo que los esfuerzos de validación se enfoquen en los Parámetros de Proceso (PPC) más Críticos.
- Atributos críticos de calidad (CQA) y parámetros críticos de proceso (CPP): las características específicas y definidas del producto (CQA) y las variables del proceso (CPP) que se deben controlar para garantizar la calidad deseada del producto, que forman los criterios de aceptación para PQ.
- Capacidad de proceso Analysis (Cpk/Ppk): el análisis estadístico realizado sobre datos de PQ para cuantificar qué tan bien está centrado un proceso dentro de sus límites de especificación y cuánta variabilidad tiene, proporcionando una puntuación numérica de su “capacidad”.
- Verificación continua del proceso (CPV): la moderna “Etapa 3” de la validación del proceso, que implica el monitoreo continuo de los parámetros del proceso y los atributos de calidad durante la producción de rutina para garantizar que el proceso permanezca en un estado constante de control.
- Gestión del control de cambios: el procedimiento formal del sistema de calidad para evaluar, documentar y aprobar cualquier cambio propuesto a un sistema o proceso validado para garantizar que no salga involuntariamente de su estado validado.
- Gestión de desviaciones y CAPA: El sistema para investigar, determinar la causa raíz e implementar acciones correctivas y preventivas para cualquier evento inesperado. eventos o fallas que ocurren durante o después de la validación.
- Validación retrospectiva: un enfoque de validación obsoleto, ahora rara vez aceptable, basado en el análisis de datos de producción históricos de un proceso existente no validado para intentar demostrar que ha estado funcionando en un estado de control.
- Informe de resumen de validación (VSR): el documento final y concluyente que resume todo el esfuerzo de validación (IQ OQ PQ), presenta los resultados clave, aborda cualquier desviación y declara formalmente que el sistema o proceso está validado y es apto para uso comercial.
Enlaces externos sobre la validación del proceso IQ OQ PQ
Normas internacionales
(Pase el cursor sobre el enlace para ver nuestra descripción del contenido)
Glosario de términos utilizados
American Society for Testing and Materials (ASTM): una organización internacional de normalización que desarrolla y publica normas técnicas de consenso voluntario para materiales, productos, sistemas y servicios, destinadas a mejorar la calidad y la seguridad en diversas industrias.
Calculation of Process Capability (Cpk): una medida estadística que evalúa la capacidad de un proceso para producir resultados dentro de límites específicos, calculada evaluando la distancia entre la media del proceso y el límite de especificación más cercano, normalizado por la desviación estándar del proceso.
Computer Numerically Controlled (CNC): un proceso de fabricación que utiliza software informático programado para controlar máquinas herramienta, lo que permite un funcionamiento preciso y automatizado de tareas como cortar, fresar, taladrar y grabar materiales.
Continuous Integration/Continuous Deployment (CI/CD): una práctica de desarrollo de software que automatiza la integración de cambios de código y la implementación en entornos de producción, lo que permite actualizaciones frecuentes, entregas más rápidas y una mejor colaboración entre los equipos de desarrollo y operaciones a través de pruebas y monitoreo automatizados.
Corrective Action and Preventative Action (CAPA): un enfoque sistemático para identificar, investigar y abordar no conformidades y problemas potenciales para prevenir su recurrencia y garantizar el cumplimiento de las normas regulatorias en los sistemas de gestión de calidad.
Critical Control Points (CCP): Etapas específicas de un proceso donde se puede aplicar control para prevenir, eliminar o reducir los riesgos para la inocuidad alimentaria a niveles aceptables. Identificar estos puntos es esencial para un análisis de riesgos eficaz y la gestión de controles críticos en los sistemas de producción de alimentos.
current Good Manufacturing Practice (cGMP): un sistema que garantiza que los productos se produzcan y controlen de manera consistente de acuerdo con estándares de calidad, abarcando regulaciones y pautas para procesos de fabricación, instalaciones, equipos y personal para garantizar la seguridad, la calidad y la eficacia en las industrias farmacéutica, alimentaria y otras industrias reguladas.
Defects Per Million Opportunities (DPMO): una medida utilizada en control de calidad que cuantifica el número de defectos en un proceso por cada millón de oportunidades de error, calculada dividiendo el número de defectos por el número total de oportunidades y multiplicando por un millón.
Define Measure Analyze Improve Control (DMAIC): una estrategia de calidad basada en datos utilizada en Six Sigma para la mejora de procesos, que consta de cinco fases: identificar el problema, medir el rendimiento actual, analizar los datos para identificar las causas, mejorar los procesos en función de los hallazgos y controlar el rendimiento futuro para sostener las mejoras.
Design Validation (DV): un proceso para garantizar que un producto cumple con los requisitos específicos y el uso previsto mediante pruebas y evaluaciones, confirmando que el diseño cumple su propósito y funciona correctamente en condiciones del mundo real.
Failure Mode and Effects Analysis (FMEA): un método sistemático para evaluar modos de falla potenciales dentro de un sistema, proceso o producto, evaluar sus efectos sobre el desempeño y priorizar los riesgos para mejorar la confiabilidad y la seguridad a través de acciones correctivas.
Food and Drug Administration (FDA): una agencia federal del Departamento de Salud y Servicios Humanos de los Estados Unidos responsable de regular la seguridad alimentaria, los productos farmacéuticos, los dispositivos médicos, los cosméticos y los productos de tabaco para garantizar la salud y la seguridad públicas a través de la evaluación científica y la aplicación de las normas de cumplimiento.
Good Manufacturing Practice (GMP): Un sistema que garantiza la producción y el control constantes de los productos según los estándares de calidad, minimizando así los riesgos inherentes a la producción farmacéutica y las industrias afines. Abarca directrices para los procesos de fabricación, las condiciones de las instalaciones, la cualificación del personal y las prácticas de documentación para garantizar la seguridad y la eficacia de los productos.
Hazard Analysis and Critical Control Points (HACCP): un enfoque sistemático de la seguridad alimentaria que identifica, evalúa y controla los peligros en puntos críticos del proceso de producción para prevenir enfermedades transmitidas por los alimentos y garantizar la seguridad del producto.
Installation Qualification (IQ): un proceso documentado para verificar que los equipos o sistemas estén instalados de acuerdo con las especificaciones, incluyendo la evaluación de los servicios públicos, las condiciones ambientales y el cumplimiento de los requisitos de diseño, garantizando la preparación para la calificación operativa.
Measurement System Analysis (MSA): un método estadístico utilizado para evaluar la exactitud, precisión y confiabilidad de los procesos e instrumentos de medición, garantizando que los datos recopilados sean válidos y consistentes para la toma de decisiones en el control de calidad y la mejora de procesos.
Operational Qualification (OQ): un proceso de validación que garantiza que los equipos o sistemas funcionen de acuerdo con requisitos específicos dentro de límites definidos, confirmando que funcionan según lo previsto en su entorno operativo.
Performance Qualification (PQ): un proceso que verifica que un sistema o equipo funciona de acuerdo con requisitos específicos en condiciones del mundo real, garantizando que realiza consistentemente su función prevista dentro de límites predeterminados.
Process Capability Index (Cpk): una medida estadística que cuantifica qué tan bien un proceso puede producir resultados dentro de límites específicos, indicando la relación entre la media del proceso y el límite de especificación más cercano, ajustado a la variabilidad del proceso.
Process Performance Index (Ppk): Medida estadística que cuantifica el cumplimiento de un proceso con los límites de especificación, calculada mediante la media y la desviación estándar del proceso. Indica la capacidad de un proceso para producir resultados dentro de los límites definidos, considerando tanto la variabilidad como el centrado.
Product Lifecycle Management (PLM): un enfoque sistemático para gestionar el ciclo de vida de un producto desde su inicio, pasando por el diseño de ingeniería y la fabricación, hasta el servicio y la eliminación, integrando personas, procesos, datos y tecnología para mejorar la calidad del producto, reducir el tiempo de comercialización y mejorar la colaboración entre las partes interesadas.
Production Part Approval Process (PPAP): un procedimiento estandarizado utilizado en la fabricación para garantizar que los proveedores cumplan con los requisitos de calidad antes de la producción en masa, que implica la documentación y validación de las especificaciones de diseño, las capacidades del proceso y las muestras de producción para confirmar el cumplimiento de las expectativas del cliente.
Quality Management System (QMS): un sistema estructurado de procesos, procedimientos y responsabilidades destinado a garantizar una calidad constante en los productos y servicios, facilitar la mejora continua y cumplir con los requisitos reglamentarios y de los clientes.
Standard Operating Procedure (SOP): un conjunto de instrucciones paso a paso creadas para ayudar a los trabajadores a realizar operaciones rutinarias de manera consistente y eficiente, garantizando el cumplimiento de las regulaciones y los estándares de calidad.
Statistical Process Control (SPC): un método de control de calidad que emplea técnicas estadísticas para supervisar y controlar un proceso, garantizando que funcione a su máximo potencial identificando variaciones y manteniendo una producción constante dentro de límites específicos.
User Requirement Specification (URS): un documento que detalla las necesidades y expectativas de los usuarios para un sistema o producto, destacando los requisitos funcionales y no funcionales para guiar el desarrollo y garantizar la alineación con los objetivos del usuario.
























Publicaciones relacionadas
Últimas publicaciones y patentes sobre grafeno
45+ Science Tricks for Games and Marketing: Data-Driven and Statistical Tricks
Uso o abuso de 25 sesgos cognitivos en el diseño y la fabricación de productos
Revised NIOSH Lifting Equation in Bench Ergonomics
Dark Web vs Darknet vs Deep Web: 101 y más
Últimas publicaciones y patentes sobre autómatas celulares